Amerindians Are Even More Genetically Diverse and Older Than You Thought
Science DOI: 10.1126/science.aab3884
Genomic evidence for the Pleistocene and recent population history of Native Americans
Raghavan, Maanasa, Matthias Steinrücken, Kelley Harris, Stephan Schiffels, Simon Rasmussen, Michael DeGiorgio, Anders Albrechtsen, …Eske Willerslev.
How and when the Americas were populated remains contentious. Using ancient and modern genome-wide data, we find that the ancestors of all present-day Native Americans, including Athabascans and Amerindians, entered the Americas as a single migration wave from Siberia no earlier than 23 thousand years ago (KYA), and after no more than 8,000-year isolation period in Beringia. Following their arrival to the Americas, ancestral Native Americans diversified into two basal genetic branches around 13 KYA, one that is now dispersed across North and South America and the other is restricted to North America. Subsequent gene flow resulted in some Native Americans sharing ancestry with present-day East Asians (including Siberians) and, more distantly, Australo-Melanesians. Putative ‘Paleoamerican’ relict populations, including the historical Mexican Pericúes and South American Fuego-Patagonians, are not directly related to modern Australo-Melanesians as suggested by the Paleoamerican Model.
Nature doi:10.1038/nature14895
Genetic evidence for two founding populations of the Americas
Skoglund, Pontus, Swapan Mallick, Maria C. Bortolini, Niru Channagiri, Tabita Hunemeier, Maria L. Petzl-Erler, Francisco M. Salzano, Nick Patterson, and David Reich.
Genetic studies have consistently indicated a single common origin of Native American groups from Central and South America. However, some morphological studies have suggested a more complex picture, whereby the northeast Asian affinities of present-day Native Americans contrast with a distinctive morphology seen in some of the earliest American skeletons, which share traits with present-day Australasians (indigenous groups in Australia, Melanesia, and island Southeast Asia). Here we analyse genome-wide data to show that some Amazonian Native Americans descend partly from a Native American founding population that carried ancestry more closely related to indigenous Australians, New Guineans and Andaman Islanders than to any present-day Eurasians or Native Americans. This signature is not present to the same extent, or at all, in present-day Northern and Central Americans or in a ~12,600-year-old Clovis-associated genome, suggesting a more diverse set of founding populations of the Americas than previously accepted.
Whole-genome and ancient DNA studies continue to topple conventional paradigms, befuddle academic researchers and fulfill out-of-America predictions. The two brand new studies by teams from the Reich lab at Harvard and the Willerslev lab at the University of Copenhagen postulate no fewer than three ancestry components in Amerindians related to three major population clusters in the Old World. Just 10 years ago the opinion was split between those scholars who imagined genetic and cultural continuity between Amerindians and East Asians and the peopling of the Americas at 15,000 years ago and those who postulated discontinuity from the ancestors of modern East Asians and the isolation of proto-Amerindians for some 15,000 years in Beringia. The latter model known as the “Beringian Standstill Hypothesis” (Tamm et al. 2007) sought to explain the presence of unique genetic signatures in modern Amerindians which required time and geographic isolation from the ancestral East Asian pool to accrue and stabilize. The Yana Rhinoceros Horn site located in close proximity to the East Siberian Sea shore and dated at 30,000 YBP provided the material evidence and the lowest chronological horizon for a proto-Amerindian source population presumably locked in a northern refugium during the LGM times and waiting for the ice shield to melt before spreading into the New World. While the two mental models differed in the extent to which they allowed for discontinuity between East Asians and Amerindians, they both imagined a homogeneous East Asian gene pool yielding an even more homogeneous Amerindian population.
With the sequencing of the DNA from the Mal’ta boy located in South Siberia and dated at 24,000 YBP, this thinking proved to be false. The Mal’ta site itself was located thousands of miles south of the mouth of the Yana River. More importantly, its DNA showed affinity to modern Amerindians and West Eurasians to the exclusion of modern East Asians (Raghavan et al. 2014). So, during the LGM times distinct Amerindian ancestry was already detectable in a geographically northeast Asian sample, while East Asian ancestry was not. Contrary to the prediction of both the East Asian Continuity and the Beringian Standstill models, a distinctive Amerindian genetic signature predated a distinctive East Asian genetic signature in the heart of Siberia and its closest affinities were with modern Europeans and not East Asians. Amerindians turned out to be older, more heterogeneous and less East Asian than everybody thought. The academic community responded to this puzzling finding by postulating an extinct “Ancient Northern European” (ANE) population that admixed with an East Asian population to generate ancestors of modern Amerindians. The ANE signature was later also found west of the Urals in ancient Kostenki DNA at 36,000 YBP (Seguin-Orlando et al. 2014; also covered here) as well as across a wide range of modern European, Middle Eastern and Caucasus populations including the putative Yamnaya ancestors of Indo-European speakers (Haak et al. 2015). But the best living example of that ancient Eurasian population continue to be Amerindians. The genetic impact of ANE on West Eurasians is so significant that all of the ancient samples discovered in Europe, from La Brana foragers to Stuttgart farmers, as well as all of the modern European populations score closer to modern Amerindians than to modern East Asians or Australo-Melanesians.
Up until now, Australo-Melanesians have never been a factor in the population genetic theories of the peopling of the Americas. Whether imagined as the earliest and sovereign wave of modern humans emanating from Africa along a “southern” migratory route or an early offshoot of an East Asian population, Australo-Melanesians have always been considered too old in terms of their divergence time and too southern in their geographic distribution to play a role in the peopling of the Americas via the Bering Strait bridge. Linguists, ethnologists, folklorists and ethnomusicologists, on the other hand, have long pointed out suggestive parallels between grammatical traits, mythological motifs, rituals and musical styles and instruments between some New World regions (especially, Amazonia but also North America) and the Sahul (see more here).
Physical anthropologists, too, advanced an argument that the earliest craniological material from the Americas is closer to Australo-Melanesians than to modern Amerindians or East Asians. This observation was so consistent across their Paleoindian sample that it warranted a formalization into a theory of two large-scale migrations to the Americas: the first one followed a coastal route and brought populations related to Australo-Melanesians from the deep south of the Circumpacific region, while the second one was derived from an inland source in northeast Asia (Neves & Hubbe 2005). Historic Fuegians and Pericues from Baha California were presented as the only surviving examples of the ancient Australo-Melanesian craniological pattern in the Americas (Gonzalez-Jose et al. 2003). However, ancient mtDNA extracted from Paleoindian skulls has invariably showed close affinities to all of modern Amerindians, thus undermining claims for a two-migration scenario (see, e.g., Chatters et al. 2014). Raghavan et al. (2015) re-examined the Paleoindian, Fuegian and Pericue craniological dataset and rejected the original conclusion:
“The results of analyses based on craniometric data are, thus, highly sensitive to sample structure and the statistical approach and data filtering used. Our morphometric analyses suggest that these ancient samples are not true relicts of a distinct migration, as claimed, and hence do not support the Paleoamerican model.”
But while the Australo-Melanesian hypothesis cannot be defended using craniological material, Skoglund et al. (2015) and Raghavan et al. (2015) have now furnished whole-genome evidence for a distinct Australo-Melanesian, or Oceanic ancestry in modern Amerindians. (Note that, ironically, the craniologists failed to see the Australo-Melanesian signature in modern Amerindian skulls.) Raghavan et al. (2015) report results from their in-depth D statistic analysis of shared drift between various populations:
“We found that some American populations, including the Aleutian Islanders, Surui, and Athabascans are closer to Australo-Melanesians compared to other Native Americans, such as North American Ojibwa, Cree and Algonquin, and the South American Purepecha, Arhuaco and Wayuu (fig. S10). The Surui are, in fact, one of closest Native American populations to East Asians and Australo-Melanesians, the latter including Papuans, non-Papuan Melanesians, Solomon Islanders, and South East Asian hunter-gatherers such as Aeta.”
Two examples from their Fig. S10 can be seen below.
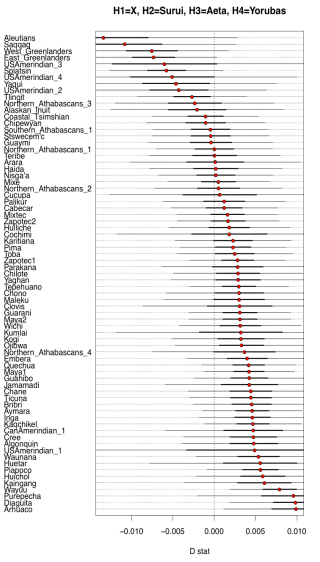
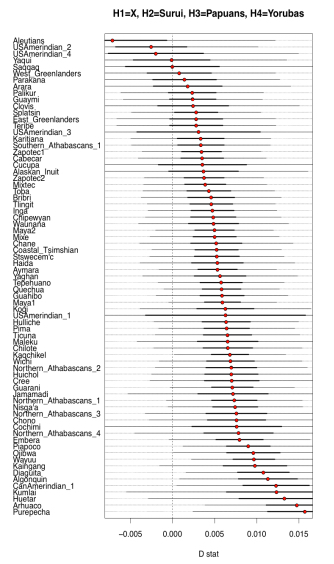
What it shows is that some Amerindian populations markedly shift in the direction of Papuans or Aeta compared to the majority of Amerindians. Importantly, this shift affects some of the same populations (e.g., Aleutians and Saqqaq) that also shift toward Han and Koryaks but, remarkably, the Australo-Melanesian pull is stronger than the East Asian pull! (see below from Fig. S10 in Raghavan et al. 2015, where negative D values are lower when Han and Koryaks are compared to Amerindians than when Papuans and Aeta are compared to them).


Importantly, the Australo-Melanesian shift is displayed by populations from both South America and North America, so it’s a low-frequency but pan-American phenomenon. Raghavan et al. (2013) showed that some North American populations are more East Asian shifted and less ANE-shifted than Central and South American populations. Now, they present evidence that those populations are more Australo-Melanesian-shifted than East Asian-shifted.
Working with a different sample, Skoglund et al. (2015) echo Raghavan et al. (2015) findings. They write:
“Andamanese Onge, Papuans, New Guineans, indigenous Australians and Mamanwa Negritos from the Philippines all share significantly more derived alleles with the Amazonians (4.6 . Z . 3.0 standard errors (s.e.) from zero). No population shares significantly more derived alleles with the Mesoamericans than with the Amazonians.”
Extended Data Table 2 in Skoglund et al. (2015) (see below) shows this excess of Australo-Melanesian alleles (Z values positive) in Amazonians (Surui, Karitiana and Xavante) compared to Central American Indians (as proxies for other Amerindians including the 12,000-year-old Anzick sample from Montana).

Interestingly, the “Australo-Melanesian” footprint in the Old World is geographically broad spanning South Asia, Southeast Asia as well as the Sahul. It’s clearly the pre-Mongoloid and pre-Austronesian “substrate” in the eastern provinces of the Old World.
Predictably, Skoglund et al. (2015) and Raghavan et al. (2015) struggled to interpret these results. Raghavan et al. (2015) concluded:
“The data presented here are consistent with a single initial migration of all Native Americans and with later gene flow from sources related to East Asians and, more distantly [this statement is contradicted by their own data, as I showed above. – G.D.], Australo-Melanesians.”
But the conclusion from Skoglund et al (2015) is radically different:
“[O]ur results suggest that the genetic ancestry of Native Americans from Central and South America cannot be due to a single pulse of migration south of the Late Pleistocene ice sheets from a homogenous source population, and instead must reflect at least two streams of migration or alternatively a long drawn out period of gene flow from a structured Beringian or Northeast Asian source. The arrival of Population Y ancestry in the Americas must in any scenario have been ancient: while Population Y shows a distant genetic affinity to Andamanese, Australian and New Guinean populations, it is not particularly closely related to any of them, suggesting that the source of population Y in Eurasia no longer exists…”
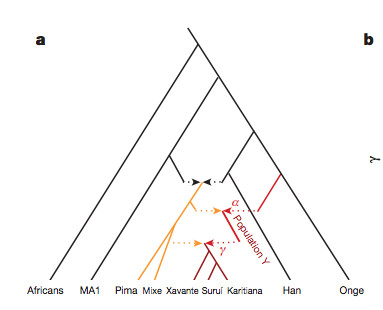
Skoglund et al. (2015) seem to be more reasonable in their judgment. Their phylogenetic tree with admixture arrows (see on the left) captures well the ever-more-complex prehistory of the New World.

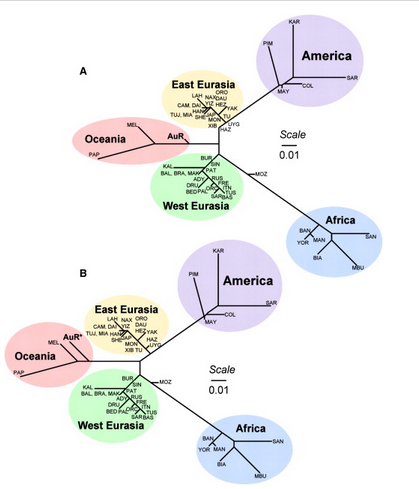
It used to be that Amerindians were depicted as a simple offshoot of a Han-like population (see the tree on the right, from McEvoy et al. 2010, Fig. 1). The branch connecting them to East Asians has always been long but it was interpreted as the effect of a bottleneck induced by the Beringian Standstill and subsequent further isolation in a newly-colonized continent. Now, Amerindian ancestry, whether northern or southern, spans the whole gamut of extra-African genetic diversity. In addition, the Australo-Melanesian link in the New World is clearly connected to the discovery of a stronger Denisovan signal in Amerindians vs. East Asians (Qin & Stoneking 2015), which suggests that Amerindians must be older than the LGM time frame entertained by Raghavan et al. (2015).
While Raghavan et al. (2015) dismiss the Australo-Melanesian hypothesis advanced by craniologists, Skoglund et al. hope that direct DNA analysis of the Paleoindian material from Amazonia will yield support to their finding. Importantly, just like the mythical “Ancient Northern Eurasian” (ANE) population to which the Mal’ta boy belonged is claimed to no longer exist in the Old World, the ancient Population Y ceased to exist, too. But apparently their descendants are well and alive in the New World.
Out-of-America can end the torturous guesswork that the population genetic community is engaged in trying to explain the high allelic diversity and the diverse set of continental connections to the Old World exhibited by Amerindian genomes. Phenomenal linguistic and cultural diversity in the Americas (with its well-documented connections to West Eurasia, East Asia and the Sahul) now receives full corroboration from population genetics. One pulse from a single, structured ancient Amerindian population followed by long-range migrations to the Sahul, Middle East/Caucasus, Europe and East Asia around 60-40,000 YBP provides an elegant explanation to the observed cross-disciplinary pattern.



[…] Anthropogenesis breaks down the recent papers on how the Americas were populated. It introduces more papers than I’m used to on proposing a European connection (via Eurasia) but the “Ancient Northern European” theory mentioned has cited papers, not entirely wild speculation, although it seems Ancient Northern Eurasians would be more accurate. […]
German,
Once again the population genetics researchers are making broad ranging statements that directly contravene the physical evidence, that archeology has provided.
As you well know, dozens sites in both north and south America, attest to a very ancient human presence in the new world. A very recent find of a disarticulated, and cracked bones mammoth, found in a fresh/salt water shellfish midden, that was several miles from the shore at the time, on the Monterey penninsula, shows that humans were there 24kya.
So I would ask both groups, if the ancestors of Native Americans were still “stuck” in Beringia until 15kya or had not arrived at all , who was killing mammoths in Cal. 62 kya or in Ok. 50kya or Co. 42kya. And who killed and butchered a mammoth and hauled saltwater shellfish, shore birds and freshwater mussels to the same pile.
By the way are you aware of the 2013? paper that pins down the surface ages ,at the Calico dig sites, to 100-198kya?
Back to an Out of America discussion, as you noted the mythological, musical ritual similarities between certain Native American and Australasian people is hard to dismiss.
From my exposure, on your blog, to these ideas I have noticed a couple of things.
The overlap of the earth diver motiff in Eurasia, with the distribution of and ritual use of Datura among the Munda speaking people.
It has recently been shown that , among a particlular culture of the US sw, the use of datura and wild tobacco are associated with rock art. Both species which are not native to the immediate vicinity of the art , are always found growing together at the base of the rocks or cliffs.
Now, here is where it gets very interesting, both of these very American species, were identified and catalogued by the easiest European naturalist to venture into the Australian bush, cir.1830’s.
They were found at inland sites across nw Australia.
The go to explanation is they were brought by ships from Mexico, but their discovery predates the arrival of such ships by several decades and were found thousands of miles from where said ships would have made port.
The species of both of these plants are most closely related north American varieties, the tobacco, is most likely that found in the Sw US, while the datura is a cousin of California’s jimsom weed.
Another important side note, the highland papuans grow a variety of wild tobacco, and have been since they were forced away from the tropical coast, by a late 13-14th century austronesian expansion.
In light of all of that I do belive, as you stated, that this so called Australasian component in native Americans is actually a native American signal in the old world.
Thanks Ron. I have to study the plants argument a little further but the Calico Hills article you referenced is likely this one: http://www.sciencedirect.com/science/article/pii/S0169555X10003703.
Did you know of this one?
J Hered (1979) 70 (5): 342-344
Interspecific hybridizations with an African tobacco, Nicotiana africana Merxm
D. U. GERSTEL, J. A. BURNS and L. G. BURK
Abstract
A recently discovered species from Africa, N. africana Merxm., was crossed as the male to 27 other species of the genus. Germinable seeds were obtained from nearly all crosses, but most seedlings died. Crosses with N.fragrans from the South Pacific, however, produced vigorous hybrids. Cytological and biochemical evidence suggest some taxonomic affinity between N. african and N. fragrans, but it remains puzzling how N. africana, or its ancestor(s), reached Africa…
Hi German, here is a link to information about a study showing human-like dexterity in wild bearded capuchin monkeys in Brazil. I don’t think directly related to this post but maybe to add to the OoAm hypothesis list of similarities between new world monkeys and humans.
Thanks for the quick analysis of these two papers. As a layperson I was a bit confused. Once again, OoAm helps make sense of emerging data while the mainstream struggles to tie it all together.
Thanks Ken. It’s a great magazine piece.
I forgot the link to the Gerstel paper-
http://jhered.oxfordjournals.org/content/70/5/342
German:
Could you explain for us clearly your tree of relationships between Amerindian and Eurasian populations? Did a population leave America, then diverge into East and West Eurasians? Did West Eurasians leave first, followed by East Eurasians? Was there further gene flow from America to Eurasia, or from Eurasia back to America, and if so at what point and between which populations?
Thanks Max. It’s good to see you signing with a “real name.” As a simplest scenario, I postulate one migration out of the Americas, with differentiation into Papuan, East Asian and West Eurasian proto-populations happening in Siberia. America’s geographic isolation and ecological complexity makes multiple migrations less likely. I interpret the various degree of relationship between Papuans and Amerindians (the weakest), West Eurasians and Amerindians (intermediate) and East Asians and Amerindians (the strongest) as a function of geographic distance from America and not of multiple migrations happening at different times. What likely happened after the original migration out of the Americas (likely during Clovis times) is Pan-American gene flow between “northern Amerindian” and “southern Amerindian” demes, as can be seen in the mostly pan-American distribution of Y-DNA hgs C and Q and mtDNA hgs C/D and A/X/B. So, if I were to draw a tree, I would split off Amerindians first (with multiple admixture edges between different subpopulations) and then create a Eurasian node followed by a long branch for Papuans, an intermediate one for West Eurasians and a shorter one for East Asians (without any migration edges between basal Eurasian populations). Does this model make sense to you?
Thank you. The one thing that is not clear is which evolutionary mechanism makes populations closer to America genetically closer to Americans if not migration – what do you have in mind?
Some West Eurasians are closer to Amerindians than Papuans are, some are further – what do you think explains this? African archaics?
MbutiPygmy Karitiana Dai Ust’-Ishim -0.1101 -23.936
MbutiPygmy Karitiana Lithuanian Ust’-Ishim -0.0362 -7.889
MbutiPygmy Karitiana Papuan Ust’-Ishim -0.0318 -6.175
MbutiPygmy Karitiana Sardinian Ust’-Ishim -0.0107 -2.309
Sorry for the ghetto edit – I mean to add that the above D-stats are of course confounded by the use of Mbuti as an outgroup, but I don’t have any using Chimp.
It’s okay, I got what you mean. Yes, IMO, Mbuti has archaic admixture, so we would need to get chimps as an outgroup for cleaner results. Genetic identity increases with decreasing geographic distance due to migration with a serial founder effect. Does it answer your question? Thanks for the stats.
Thank you.
I still seem to be missing out on an epicycle or two, because your model as I understand it still doesn’t appear to explain the data.
Using D statistics, in which branch-specific drift is cancelled out, East Asians share much more drift with Amerindians than West Eurasians do, but West Eurasians share more drift with Amerindians than they do with East Asians. In your scenario, East Asians ought to be closer to West Eurasians than they are to Amerindians, having been part of the same population for longer, or if we suppose they diverged immediately after the migration from the New World, they should at most be equidistant. West Eurasians and East Asians should be equidistant from Amerindians. The significant f4 result showing Amerindians to be closer to both West Eurasians *and* East Eurasians than they are to each other appears to violate a simple tree and require admixture (of several possible kinds, but if West and East Eurasians are a subclade of Amerindians, then gene flow from Amerindians into both groups separately would be needed.)
Keeping in mind the properties of D (and related) statistics, how do you explain the above relationships?
Good points! At some point in the past I seriously entertained a possibility of a secondary, 12,000-10,000 year old migration of Amerindians into East Asia only to explain the greater proximity between Amerindians and East Asians compared with West Eurasians. (This is still consistent with the general principle we see in the worldwide patterns of genetic variation whereby genetic distance is proportionate to geographic distance because regions that are closer geographically to each other also have a higher chance of mutual admixture.) This secondary, “Boasian” back migration would explain the sharing of such phenotypical traits as shoveling and facial flatness between Amerindians and East Asians. MA-1 is EDAR-, while a vast majority of modern Amerindians and East Asians are EDAR+, so this migration would happen in post-MA-1 times. And naturally MA-1 is free of the East Asian component, while it has one of the Amerindian one. It’s precisely northern Native Americans (EA, Na-Dene especially) that that are pulled closer to East Asians than Southern Amerindians, so the phylogeographic principle keeps working well.
Then, I dropped this scenario just to keep things simple and adhered to the phylogeoographic principle as the one that can skew such metrics as D stats. But now that you raise this issue head-on, will this secondary migration from America to east Asia only be sufficient to explain the D stats you are talking about?
A migration from Amerindians to East Asians only explains why East Asians are closer to Amerindians; it would still leave West Eurasians closer to East Asians. Another, wholly distinct migration is required, since West Eurasians with elevated affinity to Amerindians need not show any increased affinity to East Asians, and vice versa. Unless I am missing something?
Having separate ANE-like and East Asian-like American populations which mix at some point prior to Anzick-1 to form modern Amerindians ought to work, but it would require some fancy footwork to have them both independently leave America.
The Into-America scenario calls for North Asian and East Asian branches of Eurasians to meet in Beringia or thereabouts and mix together, leading to a population which shares drift with both West and East Eurasians. The location of Beringia and the restricted passage to the New World makes the meeting of separate streams of ancestry here natural, and of course they have a great deal of room in Eurasia to develop and migrate separately.
As we have discussed before, I have no objection to an argument on linguistic and perhaps cultural and archaeological grounds that the colonization of the New World must be much older than this genetic scenario posits, and that it therefore must be wrong or at least incomplete. However, you have also criticized this scenario on its own, genetic terms – could you explain what it is wrong with it strictly from the genetic angle?
Sorry, I have to ask you to rephrase the following: “A migration from Amerindians to East Asians only explains why East Asians are closer to Amerindians; it would still leave West Eurasians closer to East Asians. Another, wholly distinct migration is required, since West Eurasians with elevated affinity to Amerindians need not show any increased affinity to East Asians, and vice versa.”
I proposed one ancient “general” migration out of the Americas to Eurasia followed by a later, “Mongoloid” migration only to East Asia (without West Eurasia involved). I thought this would explain the West Eurasian-Amerindian link, on the one hand (stemming from the first migration) and the stronger East Asian-Amerindian link, on the other, stemming from the “compounding effect” of the first and second migrations. Prior to 10,000 YBP West Eurasians and East Asians were equidistant from Amerindians, but then East Asians became closer to Amerindians than West Eurasians are. What is missing from this scenario?
The problems I can see with the “admixture” of East Asians and West Eurasians prior to entry into the New World are several. 1) A biogeographic one: it’s unlikely that two radically divergent population streams originating in disparate parts of the Old World would end up mixing up in such a hard-to-access northern region; 2) Two phylogeographic objections: a) the so-called ANE component that links modern West Eurasians and Amerindians is best attested in modern Amerindians and not in modern West Eurasians; b) MA-1 is geographically an East Asian sample and no West Eurasian-proper autosomal components (WHG, EEF or EHG) are attested in the New World. Correspondingly, MA-1 is Y-DNA R and mtDNA U, which are again unattested in the Americas; 3) One populational objection: Amerindians are highly homozygous, while West Eurasians and East Asians are more heterozygous. If two populations with elevated heterozygosities indeed merged, we would have seen an uptick in heterozygosity in the resulting admixed population (or at least the maintenance of the heterozygosity levels attested in one of the two source populations). But what we see, instead, is that a supposedly admixed population, namely Amerindian, is highly homozygous. A bottleneck explanation does not work here as well as it used to when Amerindians were conceived of as a pure East Asian offshoot because if there was a dramatic founder effect followed by drift in America then how come we still see Amerindian diversity spanning both East Asian and West Eurasian alleles. As an illustrative example consider the following. If we look at Denisovans and Neandertals instead, we see that they are even more homozygous than Amerindians, hence a mixture of two “archaic” populations (or one modern, heterozygous and one ancient highly homozygous) to generate a population such as Amerindians would make sense. (Whether it happened or not I don’t know.) But two heterozygous populations coming together to create a homozygous population is unlikely. if you look at all the STRUCTURE and ADMIXTURE cluster runs done to date (e.g., Fig. SI6 in Raghavan et al. 2013), Amerindians such as Karitiana invariably unadmixed all the way up from K=2. (They are just colored first as East Asians at K=3 and then as themselves at higher levels, but there’s never a K level in which Karitiana is shown as a mix of West Eurasian BLUE and East Asian YELLOW.) Finally, a logical objection. In addition to postulating a bottleneck that dramatically reduced the heterozygosity of this presumably admixed population, while keeping the pan-European and pan-East Asian alleles intact, we have to postulate an additional migration out of East Asia to explain the D stats showing Saqqaq, Eskimo-Aleuts and Na-Dene as closer to East Asians and further removed from West Eurasians than the southern Amerindians. If an ancient admixture happened in Beringia, how come a later wave was unadmixed? Did ancient Siberians just revert to their pure East Asian state to admix into northern North Americans??!
As you can see, genetic problems with an East Asian-West Eurasian admixture in the New World are significant, if not to say insurmountable. But they become truly insurmountable if we take into consideration that recently it’s been shown that some Amerindians also share unique alleles with Papuans. Can we possibly imagine Papuans coming out of their “retirement home” in the Sahul to rush up the coast to join East Asians and West Eurasians in their panmixing fiesta of creating Amerindians?
Out-of-America disposes of all these problems. An externally unadmixed (relative to modern Old World human populations) but internally structured ancestral population maintaining long-term low eff pop size can be expected to be rather homozygous, while maintaining some allele sharing with its daughter populations that migrated into different parts of the world. I’m totally fine with Amerindian populations admixing with each other IN the New World after would-be Eurasian populations had already gone their separate ways, or with archaic hominins admixing to generate Amerindians in the Old World prior to migrating to the Americas, or both, but not with two divergent branches of modern Eurasians mixing up in Siberia to generate Amerindians.
Re: linguistic and cultural stuff. It’s precisely the striking similarity – first noticed by Renfrew – between the linguistic picture (high stock diversity in the Americas) and the genetic picture (high between-group diversity in the Americas), which is now supported by evidence of even higher between-group diversity in Neandertals and Denisovans – that makes me feel confident that we’re dealing here with one single puzzle that we haven’t cracked yet. Out of America seems to solve it all.
The devil is in the details. And I agree I need to make it work statistically. Hence, could you rephrase your objection that I highlighted at the beginning of my today’s response?
Originally East and West Eurasians would have the same affinity to Amerindians, the drift they all shared prior to the separation of Eurasians. The affinity of West Eurasians to the Amerindians admixing into East Asians is the same as their affinity to the Amerindians who stayed in the New World. West Eurasians share drift with the Amerindian portion of East Asians to the same extent as they do with Amerindians in the New World, and additional, Eurasian-specific drift with the original Eurasian portion of East Asians, so they are always going to be closer to East Asians than to Amerindians in this scenario.
As more Amerindian gene flow goes to East Asia, East Asians are drawn closer to Amerindians and further from West Eurasians, up to the extreme point where East Asians are completely replaced by Amerindians and hence are at the same distance from West Eurasians as Amerindians are. But you never can reach a point where East Asians are *further* from West Eurasians than Amerindians are, by this mechanism. You need a third, distinct migration into West Eurasians.
Now above I made the assumption that West Eurasians were equally related to both sets of Amerindians. However, this would not necessarily be the case if the migrations came from anciently diverged New World populations. Say that Eurasians split from a population P, and then further split into an East Eurasian population U and a West Eurasian population W; then a New World population N which had diverged from P earlier than Eurasians did mixed into U to form East Asian E. Afterward, N and P mixed to form modern Amerindians A. In that case W would share drift with the P portion of A that they did not share with the N portion of A and E. If the N proportion of E was sufficiently greater than the N proportion of A (it would have to be more because of the longer drift path shared by U and W), and the shared drift path of N was sufficiently long prior to the divergence of A and E, then A would share more drift with both E and W than they did with each other, explaining the relationship with only one additional migration from the New World.
However, that would mean that first population P was in Beringia and sent a branch out, then population N replaced them and sent a branch out, before P and N mixed in the New World to form A, with East Asians actually being a purer representative of N than any modern Amerindian population (in other words, the same as the mainstream scenario except that East Asians evolved in America and migrated to Eurasia rather than vice versa – population U is superfluous). You couldn’t have one of them hanging out in South America until the Clovis period. Nor could one evolve from the other, they have to be already divergent populations prior to the separation of Eurasians, and remain so until the separation of East Asians.
But even this doesn’t actually work, because West Eurasians differ in their affinity to Amerindians among themselves, independently of their affinity to East Asians. In the above scenario all West Eurasians are related to Amerindians through their original ancestry, so the shared drift remains the same regardless of how much they diverge from one another afterward. Nor can it be explained by varying degrees of archaic admixture, which would equally dilute their affinity to everyone and not just East Asians. So there still has to be an Amerindian-related component mixing into West Eurasians in varying degrees, and hence three migrations from the New World.
Perhaps you can come up with a way to reduce the number and complexity of migrations that I have not noticed. I’ll address your objections to the mainstream model later.
Thanks Max. This is good. I like how you attempt to model the process using discrete ancestral populations. While your writing your response to my critique of the mainstream model, I do want to clarify a couple of points and disagree with you a little bit. You require two migrations from America – one into East Asia and another one into West Eurasia, correct? If If I didn’t misunderstand you, this is exactly what my original model proposed. One group from a subdivided ancestral population goes to East Asia, the other one to West Eurasia. (Let’s leave PNG aside for a second.) Or do you mean that these migrations need to take place at different time intervals, rather than simultaneously?
My model of one single ancient general migration out of the Americas followed by admixture in the New World and a later gene flow from northern Amerindians into East Asians seem to account for observed patterns well. East Asians and West Eurasians don’t have to be closer to each other than either of them is to Amerindians. It would only happen if a) Amerindians, who stayed behind, did not admix with each other; b) East Asians and West Eurasians shared gene flow after their expansion from the Americas. I postulate the opposite, namely that Amerindians admixed within the New World and East Asians and West Eurasians did not admix with each other. The principle of geographic proximity correlating with genetic affinity works well here, too. West Eurasians and East Asians were progressively diverging from each other as they were colonizing the large Eurasian expanses. Amerindians, on the other hand, were geographically isolated, hence were more likely to admix with each other, as I propose they did.
D stats as well as PCAs show that all of East Asians are closer to Amerindians than they are to West Eurasians, all of West Eurasians are closer to Amerindians than they are to East Asians and all of Amerindians are closer to each other than they are to either East Asians or West Eurasians. Amerindians is a common denominator connecting all the sets. And East Asians and West Eurasians are closer to each other than either of them is to Amerindians because both of them are less Amerindian than Amerindians themselves. This is borne out by all the extant ancient DNA from the Americas: Anzick or Kenewick cluster with modern Amerindians. There’s no ancient DNA from the New World that clusters closer to either East Asians or West Eurasians.
I’d like to bring to your attention a PCA in Fu et al. (Ust-Ishim) SI S10.1B. In PC2 (SSAfricans excluded) Amerindians and Oceanians are opposed (in their own distinct way) to all other non-Africans including UI. On the vertical axis, East Asians and West Eurasians are much closer to each other than either of them is to either Amerindians or Papuans. This tells me that at 45,000 YBP Amerindians and Papuans were already diverged from what appears to be an undifferentiated Eurasian population. When we bring SSAfricans into the picture (SI S10.1A), Papuans and Amerindians are lumped with East Asians (again in line with geography) but Africans are lopsidedly skewed toward this East Eurasian cluster (on the vertical PC2 axis). If we add Neandertals and Denisovans into the mix (not shown by the Fu PCAs), then the whole Eurasian-Amerindian-Papuan cluster will be closer to those “archaics”, with Africans will be on their own (just like West Eurasians are on SI S10.1A). This tells me that Africans have strong archaic admixture (or there was massive lineage extinction outside of Africa since the Neandertal-Denisovan-human split, or both). It’s this admixture that creates an “illusion” that modern East Asians and modern West Eurasians are closer to each other than they are to modern [sic!] Africans.
To sum up it’s a pattern of allele sharing, not a pattern of allele divergence that’s a better predictor of common descent and ancestor-descendant relationships between subpopulations within a species.
There need to be three migrations altogether: an original one into Eurasia, and two more into West and East Eurasians. The timing and order of migrations isn’t fixed – except insofar as it must conform to the evidence of ancient DNA – because the different components would evolve separately in America. We need generic Eurasians by 45 000 years ago (Ust’-Ishim), the ANE-like Amerindian migration before 24 000 years ago (MA-1), and admixed Amerindians by 13 000 years ago (Anzick-1). The East Asian-like migration is not well constrained by aDNA.
In the two-migration scenario East and West Eurasians arise from separate populations within America, which later admix within America. This is a variant on the mainstream model, only instead of originating and mixing in Eurasia and migrating into America, they originate in America and separately migrate out before admixing within America. But in the mainstream model, it is particular derived branches of Eurasians which mix and migrate into America – i.e. Mal’ta Boy-like West Eurasians and Han Chinese-like East Asians. This explains why different East and West Eurasian groups differ in the amount of drift they share with Amerindians. If they originated from separate Amerindian populations, then all East and all West Eurasians would share the same amount of drift with Amerindians – East and West may differ, but not among themselves. So a third Eurasian population is required to explain the difference in affinity to Amerindians within the broader groups.
If there was ancient DNA from the New World that clustered specifically with East or West Eurasians that would be evidence that the admixture happened in the New World, as called for in your model. The mainstream model calls for admixture and drift in Northeast Asia/Beringia, leading to early Amerindians who already look close to modern Amerindians, as Anzick-1 does. (In principle the Into-America model could also have ANE and East Asia enter separately and mix in the New World, but the uniformity of ancestry in the New World beyond the Arctic argues against that – it makes more sense if the population that spread through the Americas was already mixed.)
A PCA with *one* 45 000 year old individual doesn’t tell us who was diverged 45 000 years ago. Basically he is in the middle, as you’d expect from an undifferentiated Eurasian. He is a little shifted toward Oceanians, which is also reflected in some D statistics; that could mean something. But it doesn’t mean Amerindians existed 45 000 years ago, or even that Oceanians existed 45 000 years ago.
The big V of global populations shows increasing Africanness going toward the right and increasing West Eurasianness going toward the top – not perfectly so but very close. Naturally some West Eurasian populations are pulled more toward Africans, and Africans are strung out to varying degrees along a cline toward West Eurasians, because there has been additional gene flow into and out of Africa.
The simple explanation for Eurasians being drawn toward Eurasian archaics is that they interbred with Eurasian archaics – which they did. Africans may well have mixed with African archaics also.
To return to your earlier objections:
1) I see nothing improbable about disparate streams of ancestry meeting in Northeast Asia. But suppose you are right: the Out-of-America scenario also calls for multiple populations with disparate origins entering Beringia – more of them, in fact – while giant ice sheets covered large areas of northern North America.
2a) The type specimen of ANE lived near Lake Baikal. Most North Eurasians have substantial ANE admixture. The fact that South Americans have more ANE ancestry than anyone else is exceedingly flimsy grounds for putting the origin of ANE in the New World. Should we put the origin of Fertile Crescent agriculture in Italy because Sardinians are the purest modern exemplars of Neolithic Anatolian farmers?
2b) MA-1 is indeed Northeast Asian, and geographically West Eurasian components are not found in America, though ANE does have an affinity to West Eurasians which goes back over 35 000 years:
Ust’-Ishim MA 1 Kostenki 14 African -0.0050 -5.1
Ust’-Ishim Native American Kostenki 14 African 0.0001 0.1
Ust’-Ishim East Asian Kostenki 14 African 0.0026 3.8
MA-1’s Y haplogroup diverged from R after it diverged from Q but before R1 diverged from R2, i.e. it is not part of the surviving R clade at all. Nor does his U* belong to any surviving form. His specific population probably died out though at n=1 we can hardly drawn any firm conclusions about uniparental lines.
3) A mixed population will indeed have increased heterozygosity, with later small effective population size resulting in increased homozygosity. Naturally Amerindian diversity spans both West Eurasian and East Eurasian alleles – there is no mechanism by which later drift would preferentially remove one or the other. Alleles are randomly fixed or lost at each locus regardless of their origin.
4) ADMIXTURE works by creating hypothetical ancestral populations (sets of allele frequencies) which explain as much of the variation in the data as possible (within the limitations of its algorithm). Given populations with a lot of drift specific to themselves, it prefers to make ancestral components based on those populations, because it can explain more of the variation that way. A component based on a diverse population will do only a half-assed job of explaining the variation in that population as well as in related populations. A component based on a highly drifted population will do an excellent job of explaining the variation in that population, and a half-assed job of explaining the variation in related populations. Hence, ADMIXTURE much prefers the latter option, and likes to produce components like Kalash and Druze and Karitiana.
5) The source of the additional East Asian ancestry in Siberia, Beringia, and the American Arctic would presumably be East Asia. Convenient nearby location, full of East Asians. 😉 Yes, it does require further migration from Eurasia, which fits the pattern of the MA-1 f3 heat map (SI 22.a in the Mal’ta boy paper), where ANE affinity increases as you go eastward and westward from Northern China and Eastern Siberia. Migration of an East Asian-like population from the New World, replacing pre-existing ANE-like populations, would predict a gradient from the point of origin in the New World. When exactly the East Asian ancestry spread through Siberia we don’t know yet, but preservation conditions for ancient DNA are promising. Among modern populations Paleo-Siberian speakers like Kets and Koryaks have the highest ANE (and Y hg Q), while Turkic and Tungusic speakers like Yakuts and Evenks have high East Asian (and C2 and N), like their southern cousins, so some of it may be really quite recent.
As for the Oceanian-like admixture in Amazonians, you’ll recall that it was just as close to Onge, so I very much doubt it actually came from the Sahul. It does not fit any better into Out-of-America, either – rather it calls for yet another divergence within America that migrated out to the Old World separately. I’d guess it was a peripheral East Asian population with elevated Onge-like ancestry.
Reich et al suggested that the Oceanian-like population may have entered the New World *before* the New World. This population seems to me the best candidate for a surviving genetic signal of a very early colonization of the New World.
Out-of-America doesn’t dispose of any of the problems. If modern Amerindians formed from a mixture of components in the New World, as they must have to make the model work at all, then you again will form a heterozygous mixed population which must become homozygous again by isolation. Only now it must entirely happen by drift in individual New World populations, without the addition of a Beringian founder effect.
Thanks Max. One thing I haven’t been clear about is that the one “general migration” you have in mind is the one that out-of-America posits as a founding, speciation-causing migration from the Old World to the New World. An Old World (geographically, Eurasian) hominin population spawned a proto-human population that migrated to the New World and speciated into “us.” It’s this migration that resulted in the formation of pan-human alleles now found in Africa, Papua New Guinea, East Asia, West Eurasia, i.e. everywhere. The other two-three migrations – now out-of-America as modern humans – that you envision (without any necessary chronological ordering between them unless suggested by ancient DNA) are precisely the 2-3 branches, West Eurasian, East Eurasian, Papuan of one single out of America migration to Eurasia that I posit.
This is a core clarification that I’d like to get your opinion on.
A few reactions to your individual statements are below:
You: “But in the mainstream model, it is particular derived branches of Eurasians which mix and migrate into America – i.e. Mal’ta Boy-like West Eurasians and Han Chinese-like East Asians. This explains why different East and West Eurasian groups differ in the amount of drift they share with Amerindians.”
This pattern whereby different Eurasian groups share different proportions of their ancestry with Amerindians does not seem to be relevant as an argument in favor of an Into-America scenario because Amerindian populations themselves vary between each other as well at those loci, hence we see them forming a rather long (mostly North-South) cline of their own in PCAs. So, ancestral substructure in America can explain this Old World variation just as well because it’s reflected in modern Amerindian variation.
I think you and I agree that the mainstream model, in order to work, has to explicitly show that the admixed components in the Americas are DERIVED Eurasian ones. But ANE is not a derived West Eurasian component (perfectly encapsulated by basal mtDNA U and basal Y-DNA R lineages of MA-1) and Han autosomally is as “basal” East Asian as it gets considering that they always get their own color in ADMIXTURE runs from low Ks. Tianyuan (low coverage, to be true) is “basal” to all East Asians but shares most nucleotides with Karitiana, per Fu et al. Tianyuan paper.
“A PCA with *one* 45 000 year old individual doesn’t tell us who was diverged 45 000 years ago. Basically he is in the middle, as you’d expect from an undifferentiated Eurasian. He is a little shifted toward Oceanians, which is also reflected in some D statistics; that could mean something. But it doesn’t mean Amerindians existed 45 000 years ago, or even that Oceanians existed 45 000 years ago.”
Yes and no. Yes because Ust-Ishim is indeed only one 45,000 year old individual and we need more. No, because the pattern captured in the PCA jibes well with the pattern captured by Neandertal chunk length, which shows a greater proximity of precisely Amerindians and Sahulians to Neandertals. Prufer et al. 2014 (Table S13.1) corroborates this pattern by showing a lower divergence values from the Altai Neandertal for Papuans and Amerindians. But you make a great point regarding a minor shift of UI to Papuans (compared to Amerindians!). This would mean – under an optimistic reading of such an individual case study – that UI was an incipient Papuan (in addition to a full-blown early Eurasian) who had diverged from Amerindians. The fact that we’re seeing the same “mild” Papuan signal in some modern Amerindians makes more sense now.
You: “the Out-of-America scenario also calls for multiple populations with disparate origins entering Beringia – more of them, in fact – while giant ice sheets covered large areas of northern North America.”
Not so. It’s just one hominin migration to the Americas followed by one migration of modern humans out of the Americas (with easy differentiation into regional clusters within a colonizing population) and may be another, Mongoloid one, going to East Asia only much much later, all following glacier dynamics.
You: “The fact that South Americans have more ANE ancestry than anyone else is exceedingly flimsy grounds for putting the origin of ANE in the New World. Should we put the origin of Fertile Crescent agriculture in Italy because Sardinians are the purest modern exemplars of Neolithic Anatolian farmers?”
Of course not, and I’m not saying that humans came from the Amazon Basin or Brazil. But if you’re saying that this is the pattern associated with farming expansions, I would entertain the possibility that agriculture originated in West Eurasia and not in Africa and spread to Africa from West Eurasia. And this is exactly what the recent Mota paper proposed, if I’m not mistaken.
You: “MA-1 is indeed Northeast Asian, and geographically West Eurasian components are not found in America, though ANE does have an affinity to West Eurasians which goes back over 35 000 years:
Ust’-Ishim MA 1 Kostenki 14 African -0.0050 -5.1
Ust’-Ishim Native American Kostenki 14 African 0.0001 0.1
Ust’-Ishim East Asian Kostenki 14 African 0.0026 3.8.”
I agree with you and thanks for the data pull. But if ANE originated in Europe prior to 36,000 and moved to Northeast Asia by 24,000 we would have seen Kostenki as having more ANE than MA-1. The opposite is clearly the case, which supports my contention that ANE came from Northeast Asia to Europe with the first migrants to that continent. Geography kicks in again, hence a sample that’s geographically closest to America (MA-1) has a stronger ANE (or Amerindian, in my model) signal than the one further removed but ultimately related to the former.
You: “MA-1’s Y haplogroup diverged from R after it diverged from Q but before R1 diverged from R2, i.e. it is not part of the surviving R clade at all. Nor does his U* belong to any surviving form. His specific population probably died out though at n=1 we can hardly drawn any firm conclusions about uniparental lines.”
There’s pattern whereby very ancient mtDNA and Y-DNA lineages (UI, MA-1, Tianyuan, Oase) are not part of any surviving clades. This is not necessarily because they died out, but because there may be methodological flaws in the phylogenies, which is to be expected considering that phylogenies were developed prior to ancient DNA samples becoming available. I haven’t worked with Y-DNA sequences directly, but I did notice a few of those possible flaws in current mtDNA phylogenies. They all mechanically assume that the mutation combos most frequently attested in modern samples are automatically more upstream than the mutation combos attested locally. But I did find a mutation combo attested on mtDNA hg D1g in South America that matches a mutation combo attested on all mtDNA hg C lineages (in addition to a known singleton connecting all of D1 lineages with all of C lineages), which suggests that this assumption is incorrect.
You: “A mixed population will indeed have increased heterozygosity, with later small effective population size resulting in increased homozygosity. Naturally Amerindian diversity spans both West Eurasian and East Eurasian alleles – there is no mechanism by which later drift would preferentially remove one or the other. Alleles are randomly fixed or lost at each locus regardless of their origin.”
Yes and no. Theoretically you are correct but when it comes to comparing two specific models of population evolution bound by specific data patterns, then out of America becomes more favorable than Into-America because it doesn’t require such highly demanding mental acrobatics when it comes to actual heterozygosities and such textbook platitudes as “random fixation or loss” when it comes to justifying a rather unique multipolarity of geographically isolated Amerindian genomes.
As you can see from Figure S12.1 in Fu et al. 2014, if we operate with homozygosities of more ancient Eurasian (archaic hominin) components such as Neandertal and Denisovan (and not with such recent Eurasian components as West and East Eurasian, with high heterozygosity values), we can arrive at Amerindian heterozygosities much more easily.
You: “The source of the additional East Asian ancestry in Siberia, Beringia, and the American Arctic would presumably be East Asia. Convenient nearby location, full of East Asians.”
Regrettably, East Asian Y-DNAs are mostly NO and their blood groups are mostly B – precisely the types rather poorly observed in the Americas, including the northernmost areas. So you would have to have those hypothetical East Asians expanding into the Arctic after MA-1 but without autosomal ANE but before the Holocene but without Y-DNA NO. What would those typical and populous East Asian look like genetically? And, mind you, you would need to have another bottleneck in the ANE-carrying population to explain the loss of ANE and potentially another bottleneck to explain the lack of NO in the Americas.
You: “As for the Oceanian-like admixture in Amazonians, you’ll recall that it was just as close to Onge, so I very much doubt it actually came from the Sahul…I’d guess it was a peripheral East Asian population with elevated Onge-like ancestry.”
So you, too, are compelled to locate this ancient population much north from the Sahul and much east and inland from Andaman islands. America fits this geographic expectation very well especially since it’s Amerindians that show that affinity to Onge and Sahulians and not East Asians.
You: ” If modern Amerindians formed from a mixture of components in the New World, as they must have to make the model work at all, then you again will form a heterozygous mixed population which must become homozygous again by isolation.”
Not so. If we use the even more homozygous Neandertal and Denisovan populations as a reference point for the origin of Amerindians and the rest of humans (see above), then we can see that modern humans are becoming more and more mixed (Amerindians are more than 2 times more heterozygous and admixed than Neandertals and Denisovans), more and more heterozygous and less and less isolated over time. And recent population history – the post-1492 global population expansions – supports this inference very well.
A good way to think about the difference between out of Africa and out of America is that out of Africa is very simple in the beginning (“Homo originated in Africa, Homo erectus originated in Africa and Homo sapiens originated in Africa) but gets complex and poorly understood when it comes to the last mile of H. sapiens’s journey, namely colonizing the New World. It has never been clear how continental variation (“races”) have emerged if genetically non-Africans are a “subset” of Africans. If it was that simple, we would have still been living in Africa but we are not. Out of America is complex in the beginning (a migration of a Eurasian hominin subpopulation to the New World, speciation into Homo sapiens, then migration back) but it get very easy after that as it allows for a substantial amount of continent-specific variation (amplified by local hominin admixture) with a weakening link back to the homeland in America proportionate to geographic distance from it.
Sorry, please delete the previous, it glitched.
“It’s this migration that resulted in the formation of pan-human alleles now found in Africa, Papua New Guinea, East Asia, West Eurasia, i.e. everywhere. The other two-three migrations – now out-of-America as modern humans – that you envision (without any necessary chronological ordering between them unless suggested by ancient DNA) are precisely the 2-3 branches, West Eurasian, East Eurasian, Papuan of one single out of America migration to Eurasia that I posit.”
If they are branches of one migration, then by what evolutionary mechanism did they develop different correlated allele frequencies with Amerindians? This is the sticking point. If you cannot offer a *concrete* explanation for this – not vague appeals to geographic proximity or ancestral alleles, an actual mechanism – then no one will give your model any credence.
“This pattern whereby different Eurasian groups share different proportions of their ancestry with Amerindians does not seem to be relevant as an argument in favor of an Into-America scenario because Amerindian populations themselves vary between each other as well at those loci, hence we see them forming a rather long (mostly North-South) cline of their own in PCAs. So, ancestral substructure in America can explain this Old World variation just as well because it’s reflected in modern Amerindian variation.”
Yes, Amerindians do tend to form a cline pointing toward Europe. If only there were an amply documented recent historical explanation…. 😉 But the difference among Eurasians is in any case measurable with respect to the *same* Amerindian reference population (e.g. Karitiana), so supposing that the difference in European affinity is due to ancient rather than recent structure, it as always requires that this structure was transferred in migrations out of America, hence multiple migrations from separate New World source populations are needed. Ancestral structure in America can in principle explain anything you like, there is no genetic evidence (barring lots of well-placed ancient DNA) that cannot be fit to a sufficiently contrived scenario of multiple divergent populations within the New World migrating to Eurasia.
“But if ANE originated in Europe prior to 36,000 and moved to Northeast Asia by 24,000 we would have seen Kostenki as having more ANE than MA-1.”
MA-1 has ~12 000 years of evolution shared with the population that mixed into Amerindians and *not* shared with K-14. Why on earth would *K-14* be closer to ANE? This is completely backwards.
“I haven’t worked with Y-DNA sequences directly, but I did notice a few of those possible flaws in current mtDNA phylogenies.”
mtDNA is a pain because it is so miniscule – only 16 600 bp. So it has frequent recurrent mutations and back mutations, but on the other hand they accumulate fairly slowly so the time resolution is terrible. In the coding region you have purifying selection, in the control region mutation is faster but the area is even tinier. Good luck finding the maximum likelihood tree by hand!
Y-DNA by contrast is quite well behaved, it is immensely bigger so back mutations and recurrent mutations are far rarer and more mutations accumulate so they are far better resolved in time. With full sequences Y phylogeny is crystal clear.
Ancient DNA is often included in uniparental trees, by the way. See for instance Rieux et al 2014 “Improved calibration of the human mitochondrial clock using ancient genomes” for one using many ancient samples. The Ust’-Ishim man paper built fresh trees for both mtDNA and Y-DNA.
“Regrettably, East Asian Y-DNAs are mostly NO and their blood groups are mostly B – precisely the types rather poorly observed in the Americas, including the northernmost areas. So you would have to have those hypothetical East Asians expanding into the Arctic after MA-1 but without autosomal ANE but before the Holocene but without Y-DNA NO. What would those typical and populous East Asian look like genetically?”
Really it is O that dominates in East Asia, among the Chinese C2 is just as common as N. The most recent wave looks Sakha and Tungus, for obvious reasons. Generally East Asian autosomally, Y DNA mostly N1c and C2, mt DNA mostly C and D. The more East Asian-like people who reached America that we know about from aDNA were the Paleo-Eskimos, who were about as Amerindian as Koryaks. They had some ANE, wherever they came from. Y hg Q1a1a (found also in Koryaks and Inuit and the occasional Athapaskan, with a Chinese sister branch) and mtDNA D2 (=D4e1b) – entirely D2, apparently drifted to fixation. There is also mtDNA D3 (=D4b1a) among Neo-Eskimos. Both of these are nested in Asian D4 clades – but then so is D4h3a, and Anzick-1 carried it. Unlike D4h3a though they are also found across Siberia to the Altai.
“And, mind you, you would need to have another bottleneck in the ANE-carrying population to explain the loss of ANE and potentially another bottleneck to explain the lack of NO in the Americas.”
You can’t lose ANE in a bottleneck, it’s an autosomal component, distributed across the genome. It was rather diluted by gene flow from a different population. You could certainly lose N or O in a bottleneck; the NRY is effectively a single locus, and limited to the half of the population with the most variation in reproductive success, so it is very drift-prone. But O is even now hardly to be found in Siberia, most of its clades seem to be Chinese Neolithic origin and tended to spread southward. N1c-L729 is very common but Siberian clades have quite recent TMRCAs. There is one branch that has reached West Beringia and is now common even in Siberian Eskimos, but its sister branches are mostly found in Uralic speakers from Eastern Europe, and its TMRCA is only about 3000 years ago. The older Siberian Y hgs are more likely Q1a and C2b (to which all known indigenous New World Y chromosomes belong).
“America fits this geographic expectation very well especially since it’s Amerindians that show that affinity to Onge and Sahulians and not East Asians.”
East Asians have *more* affinity to Onge and Sahulians than Americans do, as you’d expect.
MbutiPygmy Dai Papuan Ust’-Ishim -0.0423 -8.915
MbutiPygmy Karitiana Papuan Ust’-Ishim -0.0318 -6.175
The surprise was that it *varies* among Americans. Of course Papuan and Onge affinity also varies among East Eurasians, but that is expected because there has been opportunity for complex gene flow. The variation among Amerindians was unexpected because the Central and South American indigenous population was thought to have descended from one immigrant population. Descendants of that one population should not vary significantly in their affinity to Onge/Papuan, just as descendants of a single Out-of-America migration should not differ significantly in their affinity to Amerindians.
“Not so. If we use the even more homozygous Neandertal and Denisovan populations as a reference point for the origin of Amerindians and the rest of humans (see above), then we can see that modern humans are becoming more and more mixed (Amerindians are more than 2 times more heterozygous and admixed than Neandertals and Denisovans), more and more heterozygous and less and less isolated over time. And recent population history – the post-1492 global population expansions – supports this inference very well.”
As a general trend over time, due to growing populations and increased migration over greater distances, sure. Projecting this backward to the Paleolithic as some sort of rule that heterozygosity must always increase? Absolutely not.
Nor is it legitimate to use *modern* East and West Eurasians as proxies for the heterozygosity of the Paleolithic populations which contributed ANE and Northeast Asian to the New World. Modern populations are enormous and the product of extensive admixture. Small populations, such as we would expect to find in Pleistocene Northeast Asia, are highly subject to drift.
“It has never been clear how continental variation (“races”) have emerged if genetically non-Africans are a “subset” of Africans. If it was that simple, we would have still been living in Africa but we are not.”
Continental populations developed by the usual evolutionary mechanisms: drift, selection, and a limited amount of gene flow from archaic humans. No different than any other species. There is no mystery here that only Out-of-America can explain. I do not understand why you think that there is.
“If they are branches of one migration, then by what evolutionary mechanism did they develop different correlated allele frequencies with Amerindians? This is the sticking point. If you cannot offer a *concrete* explanation for this – not vague appeals to geographic proximity or ancestral alleles, an actual mechanism – then no one will give your model any credence.”
I don’t see anything particularly out of the box in my model. In the out of Africa model heterozygosity is correlated with geographic distance from Africa, which is thought to have arisen as a result of serial bottlenecks from Africa to America. I believe this correlation is misused as it a) assumes pure populations; b) ignores ancestral substructure. As a result, non-African populations, who presumably originated in Africa, cannot be shown to carry any “African” genetic signatures apart from pan-human alleles that could have arisen anywhere. But the principle of genetic distance ~ geographic distance from homeland should hold regardless of this difference in interpretation of worldwide patterns of genetic variation.
“Ancestral structure in America can in principle explain anything you like, there is no genetic evidence (barring lots of well-placed ancient DNA) that cannot be fit to a sufficiently contrived scenario of multiple divergent populations within the New World migrating to Eurasia.”
Well, I have to say “Bingo!” You admit that ancestral structure in America can explain worldwide genetic variation. I wouldn’t be dismissive of this simple insight, since Amerindian populations do show the highest degree of substructure among modern human populations (as measured by Fst and related classic metrics). African populations do not show the same level of between-group variation as Amerindians. It’s a systematic property, so we can walk on it. And it fits perfectly well with the progressive decline of the Amerindian signal as populations progressed away from America.
“MA-1 has ~12 000 years of evolution shared with the population that mixed into Amerindians and *not* shared with K-14. Why on earth would *K-14* be closer to ANE? This is completely backwards.”
I’m not understanding. If ANE originated in West Eurasia prior to MA-1 times and then migrated to America (as I thought you envision), then a 36,000 year old sample from West Eurasia should have more of that component than MA-1. No?
“Ancient DNA is often included in uniparental trees, by the way. See for instance Rieux et al 2014 “Improved calibration of the human mitochondrial clock using ancient genomes” for one using many ancient samples. The Ust’-Ishim man paper built fresh trees for both mtDNA and Y-DNA.”
Of course you can fit any sequence into a tree. This does not explain or prove that all those ancient mtDNAs were indeed lost in subsequent populations. Oase is a particularly good example: it lacks two N-specific mutations (so it looks like L0’1’2’3’4’5’6’M at those sites) but it does not have any of the African L3 mutations either. We can create another – extinct – non-African L3 lineage but that’s an easy way out. Fundamentally, this and other ancient DNA data falsifies the phylogeny. And it should because it’s direct evidence about ancient sequences. People just don’t want to establish falsification criteria and then iterate the model to make it respond to new and better data.
“With full sequences Y phylogeny is crystal clear.”
I haven’t worked withe Y sequences, so I can’t say if it is or it is not. My beef with mTDNA phylogenies is not as much narrowly about recurrent mutations as it is about how ancient, upstream clades are derived on the basis of modern data.
“N1c-L729 is very common but Siberian clades have quite recent TMRCAs.”
Wouldn’t the recency of those TMRCAs and the fact that NO are not found in the Americas mean that there’s no support from uniparental markers for a secondary migration of “pure” East Asians to northern North America suggested by scholars working with autosomal data?
“East Asians have *more* affinity to Onge and Sahulians than Americans do, as you’d expect.
MbutiPygmy Dai Papuan Ust’-Ishim -0.0423 -8.915
MbutiPygmy Karitiana Papuan Ust’-Ishim -0.0318 -6.175”
You included Dai. Can you show D stats for Han instead? Dai have an excess of Denisovan alelles in East Asia, they are more southern in their geographic distribution, so closer to PNG. You may be comparing the fraction of Papuan ancestry in Amerindians vs. Papuan-admixed Southeast Asians.
“Descendants of that one population should not vary significantly in their affinity to Onge/Papuan, just as descendants of a single Out-of-America migration should not differ significantly in their affinity to Amerindians.”
For me, it all very normal and reflects ancient modern human substructure (or archaic admixture in an African outgroup.) What matters is the fact that it’s Amerindian affinities that keep showing up in East Asia, West Eurasia and PNG.
“As a general trend over time, due to growing populations and increased migration over greater distances, sure. Projecting this backward to the Paleolithic as some sort of rule that heterozygosity must always increase? Absolutely not.”
Recall that out of Africa is built precisely on the reverse “rule” of heterozygosity decrease with progressive distance from Africa. if you reject the lawlike manner in which heterozygosity changes within a species’s history, you should reject it outright, regardless of the directionality of a postulated change. For me, I can accept the general linearity of the process as long as it’s supported by the data obtained from different time intervals and has a logical explanation. The totality of modern human genetic variation (as compared to contemporaneous chimps) is reduced, which suggests there was a founding bottleneck. And lo and behold, archaic Eurasian hominins were very homozygous, and Amerindians are the closest to them, among modern human populations, followed by Papuans. Both Papuans and Amerindians show increased autosomal affinity to Eurasian hominins, both in terms of chunk lengths and actual alleles. (Admixture here between a modern Eurasian population and an archaic population is difficult to accept because, again, this would have driven heterozygosities in Amerindians and Papuans up, plus there were no hominins in America or PNG.) The Old World is peppered with populations that fall out of the general pattern of Old World heterozygosity and approach the Amerindian-Papuan homozygosity pole: Hadza in Africa, Druze, Kalash, the Caucasus peoples, Mesolithic Loschbour – all show reduced heterozygosity compared to the bulk of Africans and Eurasians. Admixture provides a logical explanation for heterozygosity increase. Precisely due to admixture, heterozygosity in America increased from 1492 (which already falsifies the mainstream claim that Africa is the most heterozygous, hence the oldest continent!). SSAfricans and West Eurasians, between SSAfricans and archaic Africans, between East and West Africans has been proposed – all of that drives heterozygosity up.
“Nor is it legitimate to use *modern* East and West Eurasians as proxies for the heterozygosity of the Paleolithic populations…”
Ust-Ishim is as heterozygous as modern West Eurasians.
“There is no mystery here that only Out-of-America can explain. I do not understand why you think that there is.”
Not just out of America. Multiregional explains it, too. And the whole issue was raised by MR as a critique of out of Africa, and I always thought it was a valid critique.
Also re: heterozygosity vs. homozygosity. Out of Africa implies that heterozygosity was steadily increasing while modern humans were still in Africa (and that means for a good 150,000 years using current chronologies). Then, as they exited Africa, a bottleneck happened that reduced that diversity. Then another major bottleneck happened as humans colonized America. So, the heterozygosity runup in Africa lasted for a period of time that’s 3 times longer than subsequent extra-African evolutionary history. And it was not accompanied by any massive population expansions, so the cause of steady heterozygosity increase is not clear. But nobody has any problems with that. Out of America posits a heterozygosity runup for a shorter period of time and in the context of the colonization of a vast Old World landmass, which in and of itself, required demographic growth and waves of intermixing populations, not to mention the the absorption by these modern populations of archaic hominins in some parts of the Old World.
Sorry to take so long to reply, real life and all that.
The problem for your model is not so much to explain patterns of heterozygosity, which could reflect a variety of demographic scenarios, but to explain shared drift, which shows relationships between specific populations.
“Recall that out of Africa is built precisely on the reverse “rule” of heterozygosity decrease with progressive distance from Africa. If you reject the lawlike manner in which heterozygosity changes within a species’s history, you should reject it outright, regardless of the directionality of a postulated change.”
I do reject it, heterozygosity changes as a matter of demographic history – population size and gene flow. If demographic history includes bottlenecks, then heterozygosity will decline, but it is not a law that every migration or expansion must have a strong founder effect (though of course it is common). You are the one who is building a grand scheme based on heterozygosity, not I. I am looking at the relationships between specific populations.
As you say, decreasing homozygosity away from America, taken alone, *could* be explained by an expanding population mixing with diverse archaics. You may have read Pickrell and Reich’s paper last year “Toward a new history and geography of human genes” which discusses different models of Out-of-Africa expansion with the same results. Coincidentally, just last week Skoglund tweeted something to the effect that “the serial founder effect model is dead” – you can find the conversation here -> https://twitter.com/pontus_skoglund/status/656514982448877568 .
Out-of-Africa is built on more than the higher heterozygosity (and lower linkage disequilibrium and more unique alleles) within Africa, though that is one piece of evidence supporting it. The higher variance there can also be explained by in part by admixture, and as Razib Khan points out in the above thread placing the centre of diversity in *East* Africa, which has high Eurasian admixture, suggests that this is indeed a contributing factor. Furthermore, diversity is also high in South India, which again fits with higher long-term population size and a history of admixture.
BTW, why is a “run-up in heterozygosity” prior to Out-of-Africa necessary? Do we know how heterozygous African humans were 200 000 years ago?
The Into-America model postulates that Amerindians are homozygous because they have a long history of small population size. Do you have some quantitative argument for why this is insufficient to explain their observed lower homozygosity?
“If ANE originated in West Eurasia prior to MA-1 times and then migrated to America (as I thought you envision), then a 36,000 year old sample from West Eurasia should have more of that component than MA-1. No?”
No, because MA-1 is a representative of the ANE population that migrated eastward. He has common ancestors more recently with the ANE ancestors of Amerindians than with the generic West Eurasian ancestors of K-14. (Assuming the relationship is simple for the sake of argument.)
“You included Dai. Can you show D stats for Han instead? Dai have an excess of Denisovan alelles in East Asia, they are more southern in their geographic distribution, so closer to PNG. You may be comparing the fraction of Papuan ancestry in Amerindians vs. Papuan-admixed Southeast Asians.”
I don’t have D stats for Han, but Han have the same level of Denisovan as Dai. I doubt Dai are specifically Papuan-admixed, but of course they could be admixed with some Paleo-Asian population; I’d be surprised if they weren’t.
“Oase is a particularly good example: it lacks two N-specific mutations (so it looks like L0’1’2’3’4’5’6’M at those sites) but it does not have any of the African L3 mutations either. We can create another – extinct – non-African L3 lineage but that’s an easy way out. Fundamentally, this and other ancient DNA data falsifies the phylogeny. ”
What do you mean, it lacks any of the African L3 mutations? Why would it have those, if it is an early branch of N? Or do you mean it lacks the L3 mutations leading to M and N, and if so, what is your source for this? The paper includes a freshly-built tree with Oase-1, 3 other ancient genomes + a Neanderthal, and 54 modern ones, and places Oase-1 as an early branch of N with 100% support. I have not heard of any ancient DNA which falsifies the phylogeny, can you direct me to the specific evidence?
“Wouldn’t the recency of those TMRCAs and the fact that NO are not found in the Americas mean that there’s no support from uniparental markers for a secondary migration of “pure” East Asians to northern North America suggested by scholars working with autosomal data?”
No, they are represented by subclades of the East Asian uniparental markers D4 and Q1a1a, at least the Paleo-Eskimos are. There are a few other possibilities, mt hg C4c, Y hg C2b1a1, but doubtful.
“Ust-Ishim is as heterozygous as modern West Eurasians.”
I’d forgotten about that, thanks. I was thinking of Loschbour. Suggests that Ust’-Ishim’s population had not been stuck in Siberia for a long time, which makes sense as he seems to be coeval with the earliest archaeological signs of modern human presence in the area.
“The problem for your model is not so much to explain patterns of heterozygosity, which could reflect a variety of demographic scenarios, but to explain shared drift, which shows relationships between specific populations.”
Out of America explains this pattern better than out of Africa. The upstream position of Amerindians explains why it shares drift with West Eurasians (to the exclusion of geographically proximate East Asians and Papuans), East Asians (to the exclusion of West Eurasians and Papuans) and Papuans (to the exclusion of East Asians and West Eurasians). The latter link needs more work but Amerindians do show more Denisovan admixture than East Asians (http://biorxiv.org/content/early/2015/04/03/017475) and hence should be expected to be closer to Papuans as well. Amerindians also seem to share more genetic material with Neandertals (and with Neandertal-admixed ancient samples such as Oase and Ust-Ishim) than East Asians (and West Eurasians), so the data is more consistent with out-of-America than into-America. There’s nothing specific that Amerindians share with SSAfricans (to the exclusion of East Asians and West Eurasians), hence SSAfricans are likely a three-way mixture between East Asians (the Y-DNA hg D-E link), West Eurasians (Mbuti, Yoruba, etc.) and archaic Africans.
“I do reject it, heterozygosity changes as a matter of demographic history – population size and gene flow.”
I’m surprised then that you support out-of-Africa. The heterozygosity cline is a key piece of evidence for out-of-Africa. I support this cline but re-interpret it as a cline of decreasing intergroup (between-group) genetic diversity with distance from America, not decreasing intragroup (within-group) diversity from Africa to America. There could be cases your “demographic history” may complicate the linearity of the process but those needs to be argued on a case-by-case basis and not turned into a negative law whereby “demographic history always and often makes heterozygosity go up and down without any rhyme or reason.”
“You are the one who is building a grand scheme based on heterozygosity, not I.”
It’s the data pattern, not my “grand scheme” and all of out of Africa theorists give it due credit, too. We disagree on which kind of diversity (intergroup or intragroup) reflects origins not on whether the diversity cline is real.
“You may have read Pickrell and Reich’s paper last year “Toward a new history and geography of human genes” which discusses different models of Out-of-Africa expansion with the same results. Coincidentally, just last week Skoglund tweeted something to the effect that “the serial founder effect model is dead” – you can find the conversation here.”
Thanks, yes, I saw it. We’re going through a paradigm shift in population genetics and out of America makes more sense now than before. We had an nascent discussion about it 5 years ago here (http://blogs.discovermagazine.com/gnxp/2011/03/population-structure-within-africa/#.VjtxQK6rRPM) when I wrote re: John Hawks: “genetics in Africa captures the history of population admixture, not the history of population divergence.”
“The higher variance there can also be explained by in part by admixture, and as Razib Khan points out in the above thread placing the centre of diversity in *East* Africa, which has high Eurasian admixture, suggests that this is indeed a contributing factor. Furthermore, diversity is also high in South India, which again fits with higher long-term population size and a history of admixture.”
You and I agree on that. There was an early paper on genetic diversity in East Africa that postulated admixture as a better way to explain diversity there than “deep origins.” The problem is that people see the evidence disproving out of Africa but they still reaffirm it (http://www.unz.com/gnxp/fires-in-the-forest-the-revolution-in-human-evolution/). This is pseudoscience under science’s symbolic umbrella.
“BTW, why is a “run-up in heterozygosity” prior to Out-of-Africa necessary? Do we know how heterozygous African humans were 200 000 years ago?”
A bottleneck reducing the proto-modern human population to just 10,000 breeding individuals (down from some 100,000) is postulated to explain the limited genetic diversity in modern humans compared to chimps (http://www.pnas.org/content/95/4/1961.full). Higher heterozygosity in Africa is taken as a sign of Africans’ greater antiquity compared to non-Africans. So there must have been a process whereby Africans regained (or at least maintained in the face of Mid-Pleistocene demographic pressures that favored genetic drift, as we know from Neandertals and Denisovans) some of the diversity that had presumably been lost in the founding bottleneck. The logical problem with out-of-Africa is that it finds it necessary to only explain the cause of heterozygosity loss outside of Africa (bottlenecks caused by colonization) but not of heterozygosity gain in Africa. African “antiquity” compared to extra-African “recency” is not a cause for heterozygosity gain in Africa.
“The Into-America model postulates that Amerindians are homozygous because they have a long history of small population size.”
Not exactly. the Into-America model postulates a relatively late admixture between 2-3 Old World (meta, proto-) populations (West Eurasian, east Eurasian and Papuan) coupled with an even later dramatic bottleneck. Out of America postulates demographic and historical continuity between the original founding modern human (or even earlier, East Eurasian hominin) bottleneck and attested low heterozygosity in America and explains the multipolar connections between Amerindian and Old World populations as population divergence subsequent to repopulation of the Old World by now-modern humans.
“e has common ancestors more recently with the ANE ancestors of Amerindians than with the generic West Eurasian ancestors of K-14.”
Could you explain this? in all the trees that include ANE, ANE is coterminous with EHG and WHG, so even under the current models ANE is not derived from any EHG-WHG clade. And considering that Amerindians sit in both ANE and East Asian clades (but East Asians don’t sit in the ANE clade), a proto-Amerindian population should be upstream from any West Eurasian ancestor of K14.
“I don’t have D stats for Han, but Han have the same level of Denisovan as Dai.”
AFAIKS, Dai have a little more Denisovan admixture than Han. (http://biorxiv.org/content/biorxiv/suppl/2015/04/03/017475.DC1/017475-1.pdf.) And a “little” in the case of Denisovan admixture can run a long way, I suppose.
“What do you mean, it lacks any of the African L3 mutations? Why would it have those, if it is an early branch of N?”
I did mean that Oase does not have any of African-specific L3a through L3h alleles. But we do need to find a lineage with African-specific mutations outside of Africa (either in a marginal modern population such as Andamanese situated along the putative southern route of expansion out of Africa, or in Ust-Ishim, or in Oase) in order to report “evidence” for an out-of-Africa migration. It’s just a methodological “must”, which out-of-Africanists have failed to turn up. In the case of out of America, it can be supported, on the mtDNA side, precisely in the presence of Amerindian-specific markers within the A2, C1 and D4h clades at low frequencies (!) outside of America (namely in Siberia). It could be a bigger methodological problem here, as the mtDNA phylogeny is constructed in such a way that all the ancestral mutations are present on all continents and on all lineages, so the radiation could have started from anywhere.
“No, they are represented by subclades of the East Asian uniparental markers D4 and Q1a1a, at least the Paleo-Eskimos are.”
Note that those supposedly later arrivals to America represent lineages from the same clades that were already present in the Americas. So, Amerindians have D1, D4h3 as old lineages and then D2/D3 (old nomenclature?) as late arrivals. Why didn’t the late migrants bring other lineages from clades previously unattested in America? This does not make sense. And it’s precisely what we see in Y-DNA and classical markers (blood group B and Y-DNA NO didn’t make it to the Americas). An alternative is that D1, D4h3 and D2/D3 are Amerindian retentions, so I would re-do the sequence analysis to see if they share mutations with each other. Considering that D1g shares unique mutations with all of C lineages (see above), we are potentially looking at radically different phylogeny here.
I tweeted back at Pontus at https://twitter.com/pontus_skoglund/status/656514982448877568. I met him at the last AAPhAs.
I’m afraid I’m not going to have time to continue this discussion. It has been quite enjoyable.
Good luck with the radically revised mtDNA phylogeny, you will need it.
Thanks Max for all your thoughts. I enjoyed our exchanges. If you procure stats for Han instead of Dai, please send them my way.