Peruvian Amerindians Have Strongest Genetic Ties to Archaic Hominins
Molecular Biology and Evolution (advance publication, October 18, 2016)
Signatures of archaic adaptive introgression in present-day human populations
Racimo, Fernando, Davide Marnettob, and Emilia Huerta-Sánchez
Comparisons of DNA from archaic and modern humans show that these groups interbred, and in some cases received an evolutionary advantage from doing so. This process – adaptive introgression – may lead to a faster rate of adaptation than is predicted from models with mutation and selection alone. Within the last couple of years, a series of studies have identified regions of the genome that are likely examples of adaptive introgression. In many cases, once a region was ascertained as being introgressed, commonly used statistics based on both haplotype as well as allele frequency information were employed to test for positive selection. Introgression by itself, however, changes both the haplotype structure and the distribution of allele frequencies, thus confounding traditional tests for detecting positive selection. Therefore, patterns generated by introgression alone may lead to false inferences of positive selection. Here we explore models involving both introgression and positive selection to investigate the behavior of various statistics under adaptive introgression. In particular, we find that the number and allelic frequencies of sites that are uniquely shared between archaic humans and specific present-day populations are particularly useful for detecting adaptive introgression. We then examine the 1000 Genomes dataset to characterize the landscape of uniquely shared archaic alleles in human populations. Finally, we identify regions that were likely subject to adaptive introgression and discuss some of the most promising candidate genes located in these regions
I haven’t been blogging that much lately. And for a good reason. It’s time to wait and absorb. Population genetics is undergoing a revolution due to influx of ancient DNA samples and whole-genome sequencing. Our data is becoming more directly related to the questions we are trying to answer. And the questions we should be asking are evolving with it.
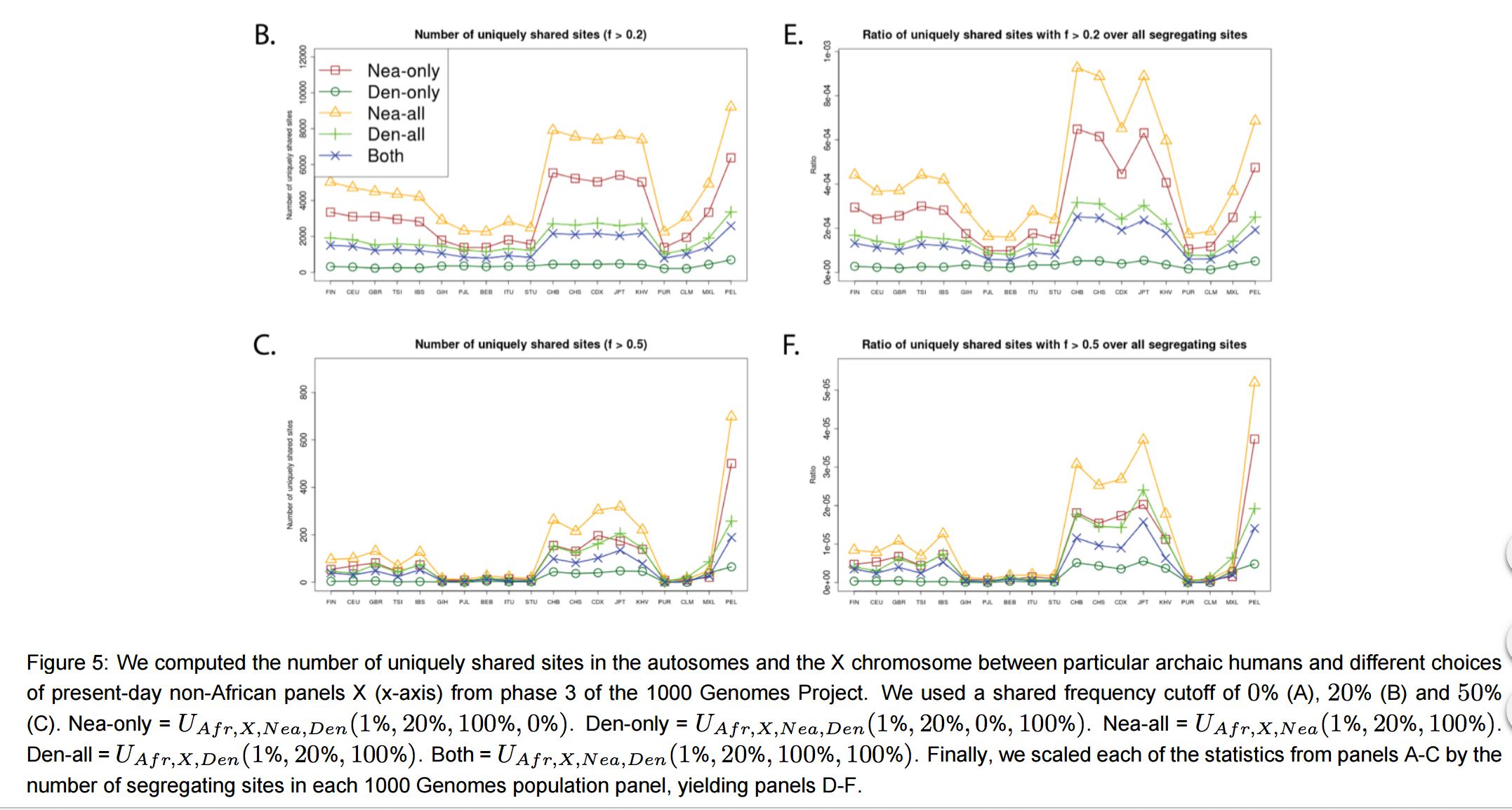
A new paper on “archaic introgression” in Eurasians and Amerindians by Racimo et al. (2016) has struck me as the one that furnished one more piece of direct evidence for the out-of-America II scenario of human evolution that I believe has a lot of potential in explaining the origin of present-day genetic, biological, linguistic and cultural distributions. Racimo et al. (2016) identified native Peruvians as holding a significant excess in direct matches with Neandertals and Denisovans compared to Eurasians (East Asians, South Asians and Europeans).
“Surprisingly, the Peruvians (PEL) harbor the largest amount of high frequency mutations of archaic origin than any other single population, especially when using Neanderthals as bait (Figures 5.B-C,S21). …Scaling the uniquely shared sites by the total number of segregating sites per population panel mitigates (but does not completely erase) this pattern. After scaling, PEL shows levels of archaic allele sharing within the range of the East Asian populations at x
= 20% (Figure 5.E), but is still the panel with the largest number of archaic sites at x = 50% (Figure 5.F).”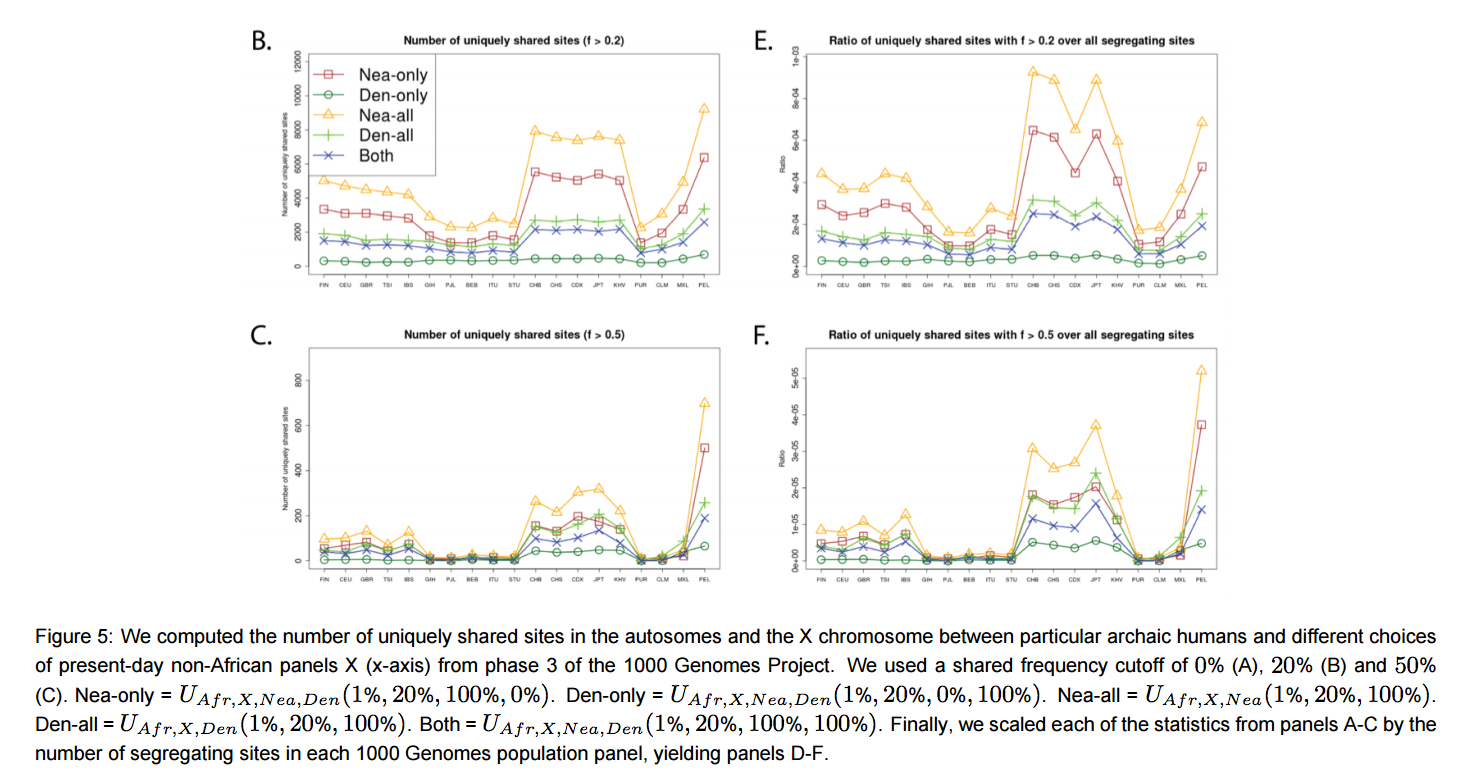
Racimo et al (2016) continued their tests and observed the same effect again:
“Furthermore, we plotted the values of UAF R,X,Nea,Den(1%, x, y, z) and Q95AF R,X,Nea,Den(1%, y, z) jointly for each population X, under different frequency cutoffs x. When x = 0%, there is a generally inversely proportional relationship between the two scores (Figure S23), but this becomes a directly proportional relationship when x = 20% (Figure 6) or x = 50% (Figure S27). Here, we also clearly observe that PEL is an extreme panel with respect to both the number and frequency of archaic shared derived alleles, and that East Asian and American populations have more high-frequency archaic shared alleles than Europeans.”
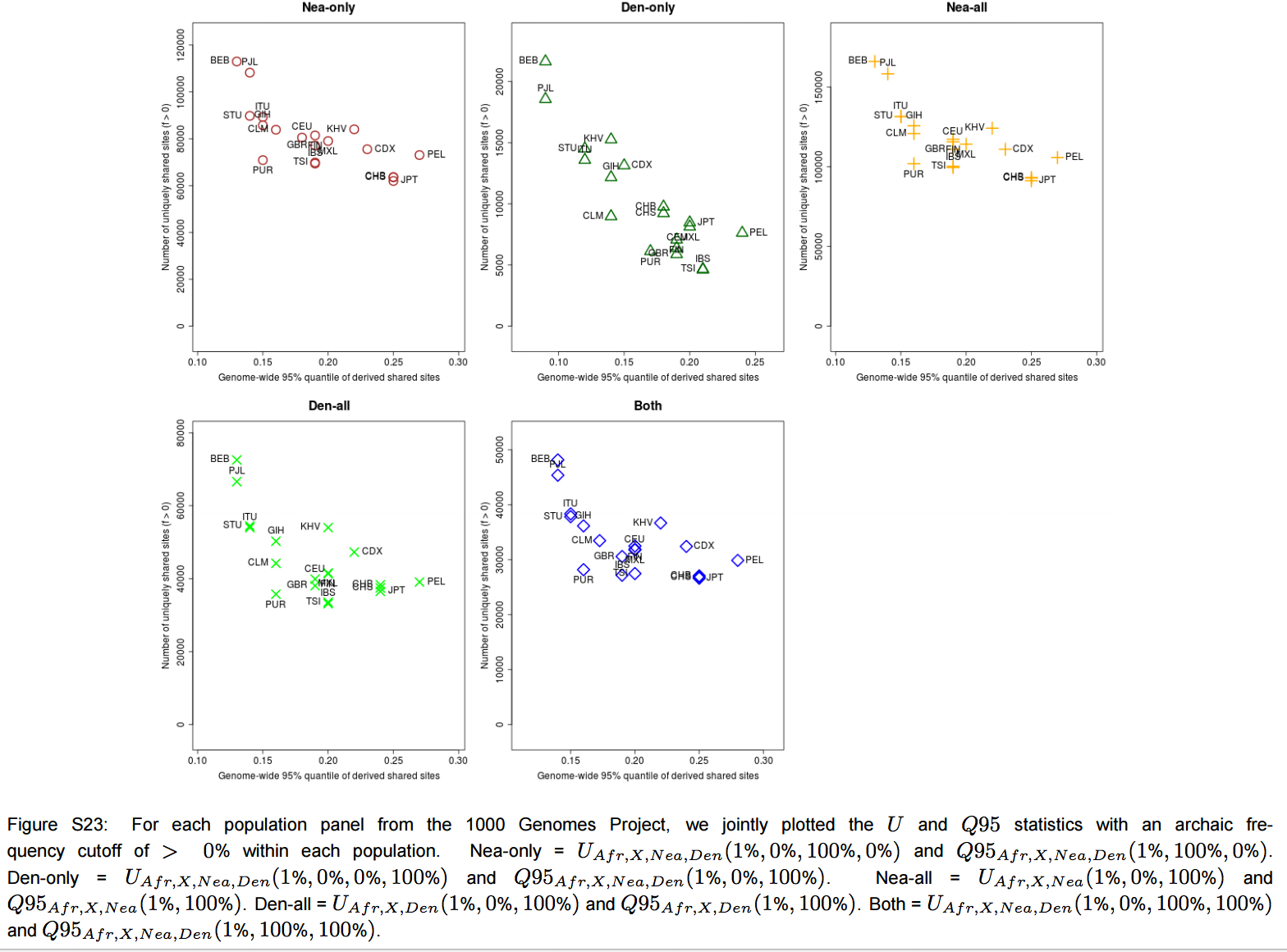
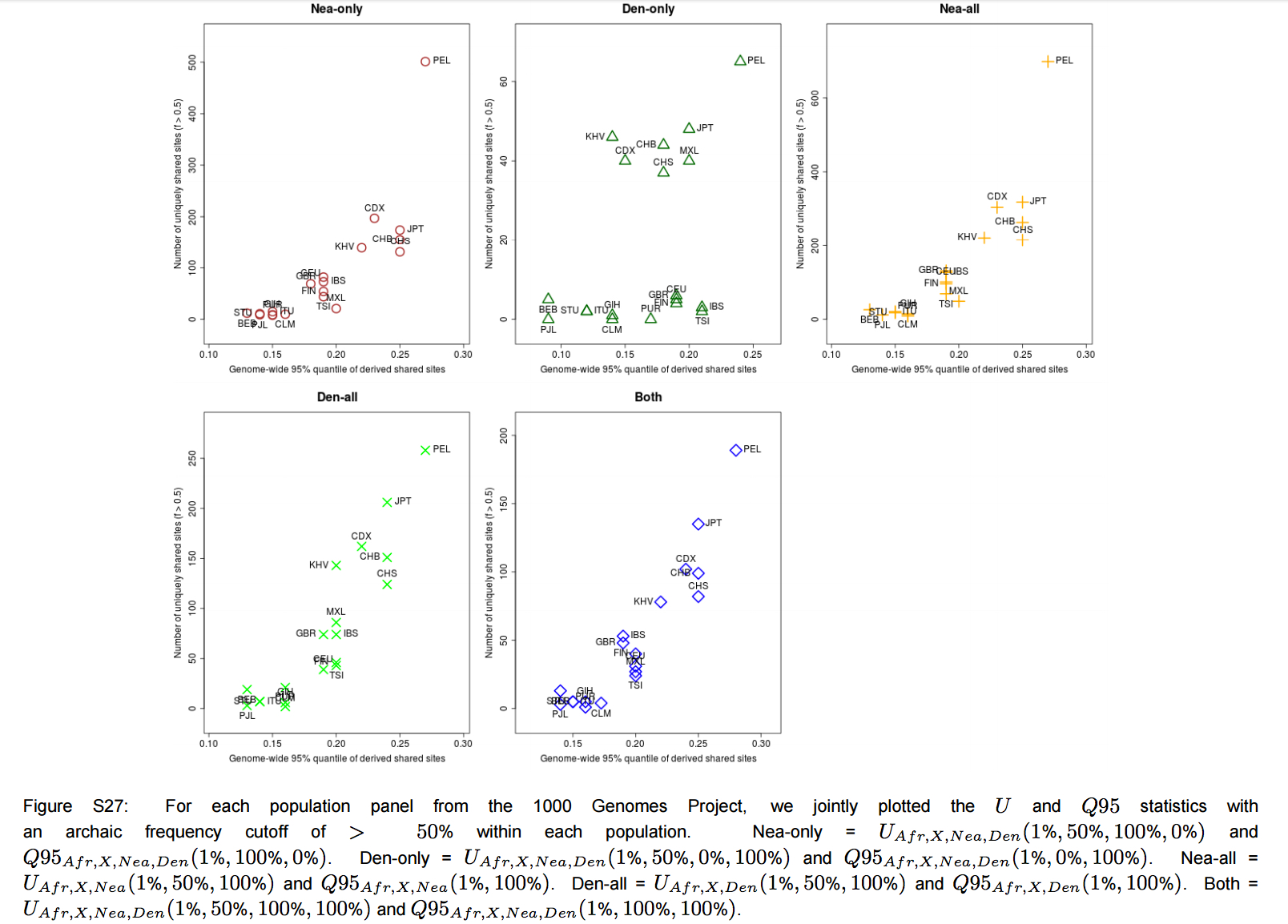
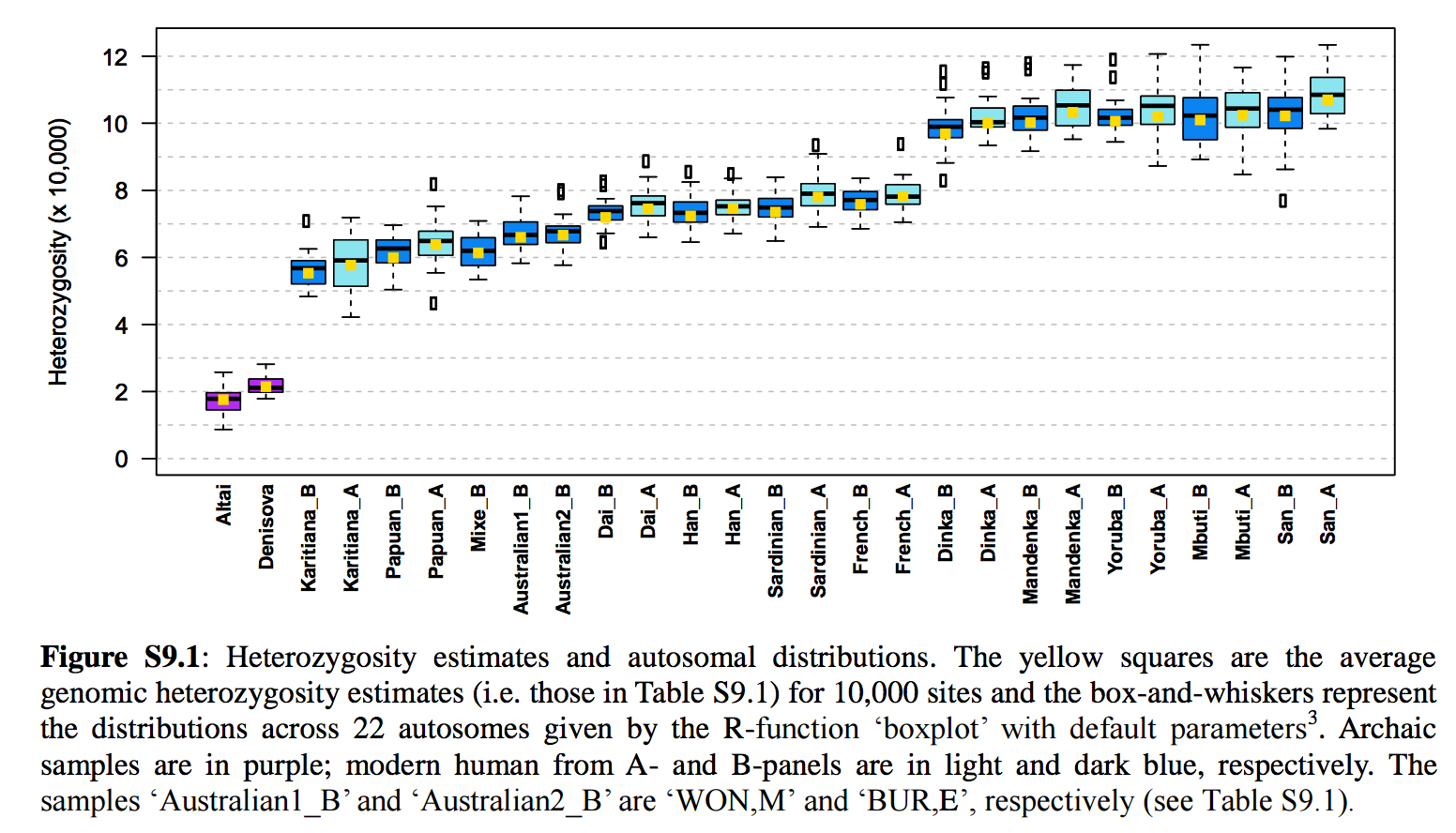
We have seen this pattern multiple times already. Karitiana were shown to possess the longest chunks of Neandertal DNA detected in the 45,000-year-old Ust’-Ishim sample than any Eurasian population. Prufer et al. 2014 (Table S13.1) documented a lower divergence values from the Altai Neandertal for Papuans and Amerindians. This makes all the more sense if we take any of the worldwide PCAs based on a whole-genome analysis(see below, from Vernot et al., “Excavating Neandertal and Denisovan DNA from the genomes of Melanesian individuals,” Science, 2016, Fig. 1) and observe that at PC-1 Amerindians (look from left to right below) are the most divergent population from Sub-Saharan Africans.
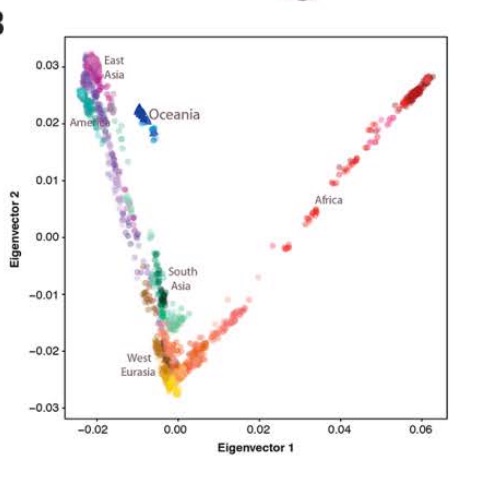
Since Sub-Saharan Africans have the least Neandertal and Denisovan “admixture” of all modern human populations, an excess in archaic alleles would pull Amerindians the furthest from them.
Racimo et al. (2016) note that Peruvians (in a panel of 1000 genomes) are the “purest” Amerindians. They didn’t report any archaic sites shared between Amerindian and Europeans to the exclusion of East Asians (in refutation of the often made claim that Amerindians are a cross between East Asians and West Eurasians), although there are archaic sites that link Amerindians and Melanesians (e.g., the genes FAP/IFIH1 coding for Type 1 diabetes). When they attempt at an explanation of the observed anomaly in the worldwide distribution of hominin ancestry, they land in a familiar territory:
“Indeed, population structure analyses of the 1000 Genomes samples suggest that Peruvians have the largest amount of Native American ancestry and show a bottleneck with a lack of recent population growth, which could explain this pattern. We also observe an increase in the variance of the distribution of U and Q95 in the presence of a bottleneck, especially when long and severe.”
What Racimo et al. (2016) have failed to bring up is the highly important pattern whereby Neandertals and Denisovans show signs of an even more severe bottleneck than Amerindians. This can be seen in the following graph from Prufer et al. (2014):
If Amerindians are closer to archaic hominins in terms of the actual shared sites and in terms of their population structure, then the crossing of the Bering Strait by would-be Amerindians must have occurred at a time when archaic hominins were still around, at a time when populations were still small, highly mobile and mutually isolated, and at a time when no other modern humans were around. It’s only after Amerindians had branched off, a backflow of emerging modern human populations into the Old World began: they were progressively losing traces of their Eurasian archaic ancestry as they moved from East Asia to Europe and Africa and they were growing in size radiating to the four corners of the world. Amerindians did not revert back to Mid-Pleistocene Population structure 15,000 years ago to selectively retain Old World hominin ancestry in the New World (this would be a very odd scenario). They have maintained the structure and the genetic material during the tens of thousands of years of isolation in the New World.



{kind=link}
Glad to see you back commenting German,
In the couple of years since I discovered your blog, I have tried to find older work by Carter and McNeish, that detailed a group of sites that were too old to be accepted at the time and they both found similar pebble tool lithics on the west coast. Carter even describes how the Dieguito(pebble tool) culture was interrupted during the YD, by an intrusive bifacial blade complex( the western most expression of Clovis related people), for several hundred years before reverting back to a pebble tool/unifacial chopper tradtion. Could that tool tradition be a hold over from earlier archaics?
Great information! Thanks.
Glad to see this bog active again.
I have noted anthropological traits among the Andean Incas that correspond closely to traits among the ancient Nilotes in the R Haplogroup.
This may get things totally wrong, but most of what I read about DNA roots traces everything back to Africa. This seems to be the totally accepted dogma by mainstream thinkers. On the other hand are great arguments like the ones above, dealing with other issues, and ancient hominids, but doesn’t this still leave the argument that mtDNA and YDNA clades found in the New World are subordinate to those from Eurasia?
I personally think that modern man rose when homo erectus or homo hidelbergensis mated with a Neanderthal or Denisovian and something happened in the brain enabling the telling of stories and language which in turn enables culture. I wait for the day when an intact 70,000 year old skeleton of a modern human is found in the state of Washington or somewhere. In the meantime I am about to publish a novel, Strong Heart by Iron Twine Press, March 2017, which is really a coming of age quest tale but seeks to show how the Native Americans have it right – they “have always been here.”
German if you have the time and interest I’d love to send you an early copy to read, get your reaction…..
Thanks Charlie. I would like to read your novel.
I too am enjoying the articles and conversations on this site.
I live in Southeast Alabama where I find stone tools on my property that look very similar to Levoillois found in western Europe.
Mr. Sheldon and I have had similar ideas that the Native American people who say they have always been here are correct. I would enjoy reading his book as well.