World Science en Route from Out-of-Africa to Out-of-America: First Stop is Out-of-Asia
bioRxiv
, Dejian, , , , , , and
Modern Human Origins: Multiregional Evolution of Autosomes and East Asia Origin of Y and mtDNA
Recent studies have established that genetic diversities are mostly maintained by selection, therefore rendering the present molecular model of human origins untenable. Using improved methods and public data, we have revisited human evolution and derived an age of 1.91-1.96 million years for the first split in modern human autosomes. We found evidence of modern Y and mtDNA originating in East Asia and dispersing via hybridization with archaic humans. Neanderthals and Denisovans were archaic Africans with Eurasian admixtures and ancestors of South Asia Negritos and Aboriginal Australians. Verifying our model, we found more ancestry of Southern Chinese from Hunan in Africans relative to other East Asian groups examined. These results suggest multiregional evolution of autosomes and East Asia origin of Y and mtDNA, thereby leading to a coherent account of modern human origins.
It appears that world science is leaving out-of-African behind. First, aDNA from Neanderthals and Denisovans has falsified the strongest form of put-of-Africa thinking, the one that postulated the complete replacement on Eurasian hominins by the incoming modern humans. Now, a Chinese team has advanced an out-of-Asia theory for modern human origins based on Y-DNA and mtDNA and revived the Multiregional Evolution model based on autosomal evidence. They’ve provided the first independent support for my long-standing critique of the out-of-Africa interpretation of mtDNA and Y-DNA phylogenetic trees. Yuan et al. (2017) are the first (since Johnson et al. 1983) to generate trees from uniparental markers that demonstrate a Eurasian origin of African lineages. Even the confused and chaotic critique of out-of-Africa from Y-DNA standpoint by Russia’s patriotic scientist, Anatole Klyosov (see my coverage of his feud with Russia’s official scientists), stopped short of developing a robust alternative tree. Yuan et al.’s major revision of the scientific theory modern human dispersals makes a critical step toward harmonizing signals coming from different molecular systems as well as harmonizing the totality of molecular evidence with evidence coming from worldwide language family distributions and the distributions of cultural traits.
Yuan et al. (2017) is currently under peer review and discussions around it are happening here and here. Pseudoanthropological blogs such as Dienekes, Eurogenes, RazibKhan, Dispatches and ForWhat are keeping notorious silence. Serious platforms such as Anthropology.net, Sarkoboros and John Hawks’s blog are silent, too. The main Russian portal for human evolution and physical anthropology is too busy popularizing science and celebrating the advantages of science over pseudoscience to timely respond to any significant developments in the field. Its antithesis, Pereformat.ru, a safe haven for Anatole Klyosov and his team of Russian patriotic thinkers, has not reacted either.
The Chinese team is led by Shi Huang, a US-trained geneticist, epigeneticist and the author of the “maximum genetic diversity” (MGD) theory of evolution. Yuan et al. (2017) draw on MGD in some of their interpretations of modern human genetic diversity. For instance, according to MGD, there are functional constraints on genetic diversity growth. This confounds phylogenetic inferences between and within species because of homoplasies caused by mutational saturation in fast-evolving parts of the genome. I will dedicate separate blog entries to MGD in the near future. For now, let’s review the out-of-Asia model of modern human origins and dispersals. Similarities between my thinking (as expressed on this blog, e.g., here, and in discussion forums around the web) and some of the Yuan et al. ideas are remarkable and coincidental. They result from looking at the same facts and making the same direct observations from those facts, not from direct collaboration, which makes them even more valuable.
Yuan et al. (2017) confirm that “genetic diversity” (as measured by pairwise sequence difference) is world-highest in Africans and world-lowest in Amerindians. They also confirm that Africans and Amerindians are genetically most divergent from each other (see below).

Africans are also found to be closer to each other than they are to other groups. This conclusion the authors specifically call out in their paper. What they don’t call out but what seems to be present in their graph above is that Amerindians are more internally differentiated than other continental groups. New World subpopulations are more distinct from each other than other continental groups’ subpopulations are distinct from each other. This fits with other metrics (such as Fst) showing that Amerindians have world-highest values of intergroup diversity (see, e.g., here and here).
Where Yuan et al. (2017) diverge from consensus is when they argue that African genetic diversity is not a function of greater antiquity compared to the rest of human continental groups but a product of selection. This is supported by the data showing increased African diversity in all of the non-neutral SNPs tested by the authors (see below).

In agreement with my critique of genetic evidence for out-of-Africa (e.g., here), they also note that higher heterozygosity can be explained as resulting from admixture, as modern admixed populations such as MXL and PUR in their sample amply demonstrate. After additional tests and with hints from MGD theory, Yuan et al (2017) propose an important methodological rule for building phylogenetic trees and inferring dates of molecular divergence:
“only hom distance [distance between homozygous alleles only. – G.D.] calculated from the slow nonsyn SNPs, hereafter referred as the slow SNPs, can be informative to phylogenetic inferences.”
In a major departure from more mainstream evolutionary theory, MGD posits that chimps are closer to gorillas and orangutans than they are to humans (see Huang, S. “Primate phylogeny: Molecular evidence for a pongid clade excluding humans and a prosimian clade containing tarsiers.” Sci China Life Sci 55 (2012): 709-25). Using 17.6 myr as a divergence time between humans and Pongidae, Yuan et al.’s (2017) estimated molecular divergence between the autosomes of major human continental groups (as derived from Esen in West Africa and British in the U.K.) to be 1.96 myr or 1.91 myr (as derived from the distances between Esen and Southern Han). This is consistent with paleobiological evidence for a Homo migration out of Africa ~ 2myr.
Yuan et al. (2017) use this autosomal molecular divergence dates as evidence for Multiregional Evolution. They believe that there were two out-of-Africa migrations: one involving Homo erectus some 2 myr ago and the other one involving the ancestors of Neanderthals and Denisovans who left Africa roughly 1.5 myr after the divergence of the Homo clade. They dispute the argument made over and over again by other genetic labs that modern humans absorbed genetic admixture from such Eurasian hominins as Neanderthals and Denisovans. This argument was based on an observation of a set of derived (as compared to a reference chimpanzee sequence) autosomal markers that are shared between non-Africans, on the one hand, and/or Neanderthals and Denisovans, on the other. In all those cases, modern Africans agree with chimpanzees in showing ancestral alleles. Yuan et al. (2017) respond:
“The assumption of D-statistics is that all modern groups are equidistant to chimpanzees so that presence of derived alleles (different from chimpanzees) was due to gene flow from Neanderthal. If in fact Africans are closer to chimpanzees or carrying more ancestral alleles in general, the conclusion of gene flow into non-Africans would become invalid. We examined this by measuring genetic distance between 1000 genomes and 10 previously sequenced chimpanzee genomes (de Manuel et al., 2016). Using the random 255K SNPs set, we found closer hom distance between Africans and chimpanzees than between non-Africans and chimpanzees (Supplementary Figure S5). As presence of Neanderthal derived alleles in a non-African are mostly in het state (Fu et al., 2014), which could be observed to be biased toward non-Africans only if Africans are in hom ancestral state, the fact of more hom ancestral alleles in Africans (or closer hom distance between Africans and chimpanzees) therefore deems invalid the previous finding of Neanderthal gene flow into non-Africans.”
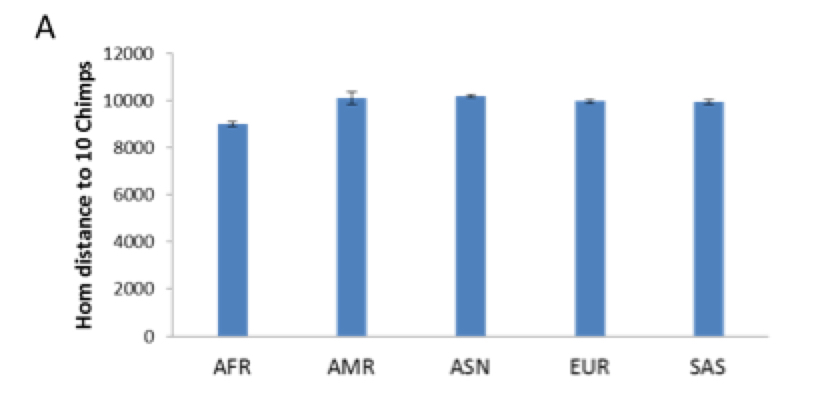
Even a random set of SNPs sometimes shows the same result (see below, from Sikora et al. “Population Genomic Analysis of Ancient and Modern Genomes Yields New Insights into the Genetic Ancestry of the Tyrolean Iceman and the Genetic Structure of Europe.” PLoS Genetics 10 (5): (2014) e1004353, Fig. S9)
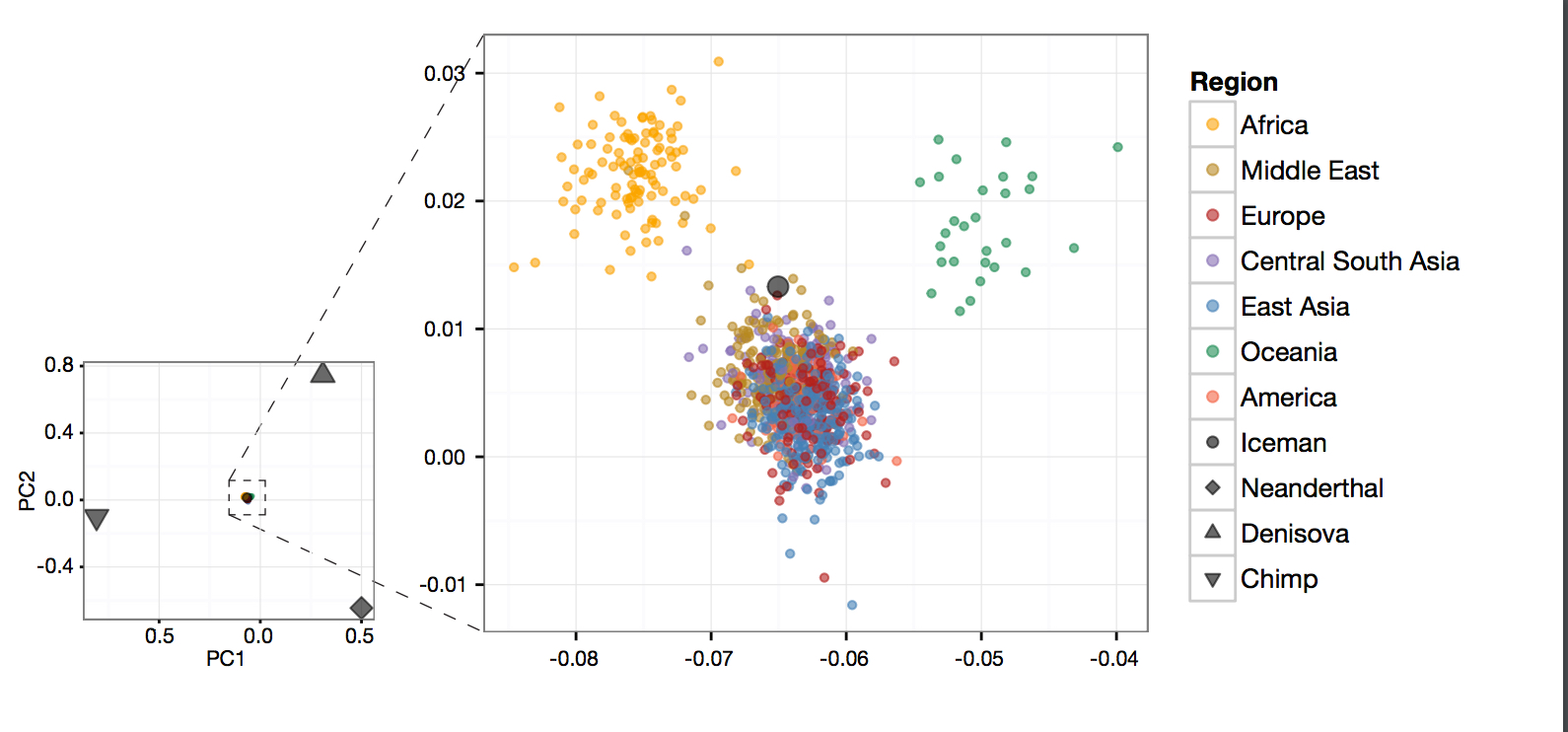
In addition, the MGD theory questions the reliability of ancestral vs. derived state assignment because of the increased probability of homoplasies in mutationally saturated genomes. But, intriguingly, Yuan et al. (2017) found modern Africans to be generally closer to Neanderthals and, especially, Denisovans (both Africa-derived) than non-Africans (see below Fig. 4A).

The authors explain this finding as evidence of dual admixture between a) non-African Homo sapiens sapiens and Africa-derived Neanderthals and Denisovans and b) between modern Africans and African hominins, the collateral relatives of Neanderthals and Denisovans who stayed back in Africa. According to Yuan et al. (2017), uniparental markers cast a light on the ultimate geographic origin of Homo sapiens sapiens that predated the admixture with the hominins. They reinterpret Y-DNA and mtDNA phylogenetic evidence as supporting a single Out-of-Asia origin for Homo sapiens sapiens.
One of the implications of the MGD theory is that only slow-evolving sites can be used for building phylogenies. Yuan et al. (2016) repeat an observation made by (Poznik et al. “Sequencing Y Chromosomes Resolves Discrepancy in Time to Common Ancestor of Males Versus Females.” Science 341, no. 6145 (2013), 562-5) that higher-order Y-DNA clades (and they tend to be African) have a lot of inconsistencies (see below, Table S6).
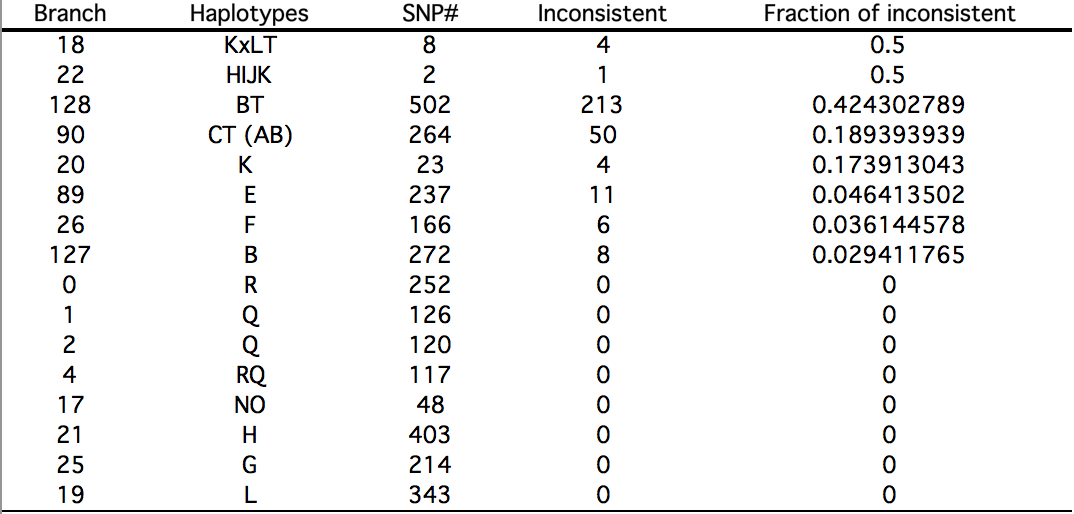
In the most glaring example, in BT clade 213 sites out of 502 are inconsistent. This means precisely what MGD predicts: unless fast-evolving sites are “winnowed out,” a tree built from a mix of slow and fast sites will have a lot of look-alike alleles that appear to be due to common descent but in reality they are just by-products of mutational saturation in a lineage of sufficient time depth.
Once Yuan et al. (2017) removed all the “noise” from the Y-DNA phylogeny, they obtained a tree that’s radically different from the canonical one (as exemplified by Poznik et al.’s tree, see below).
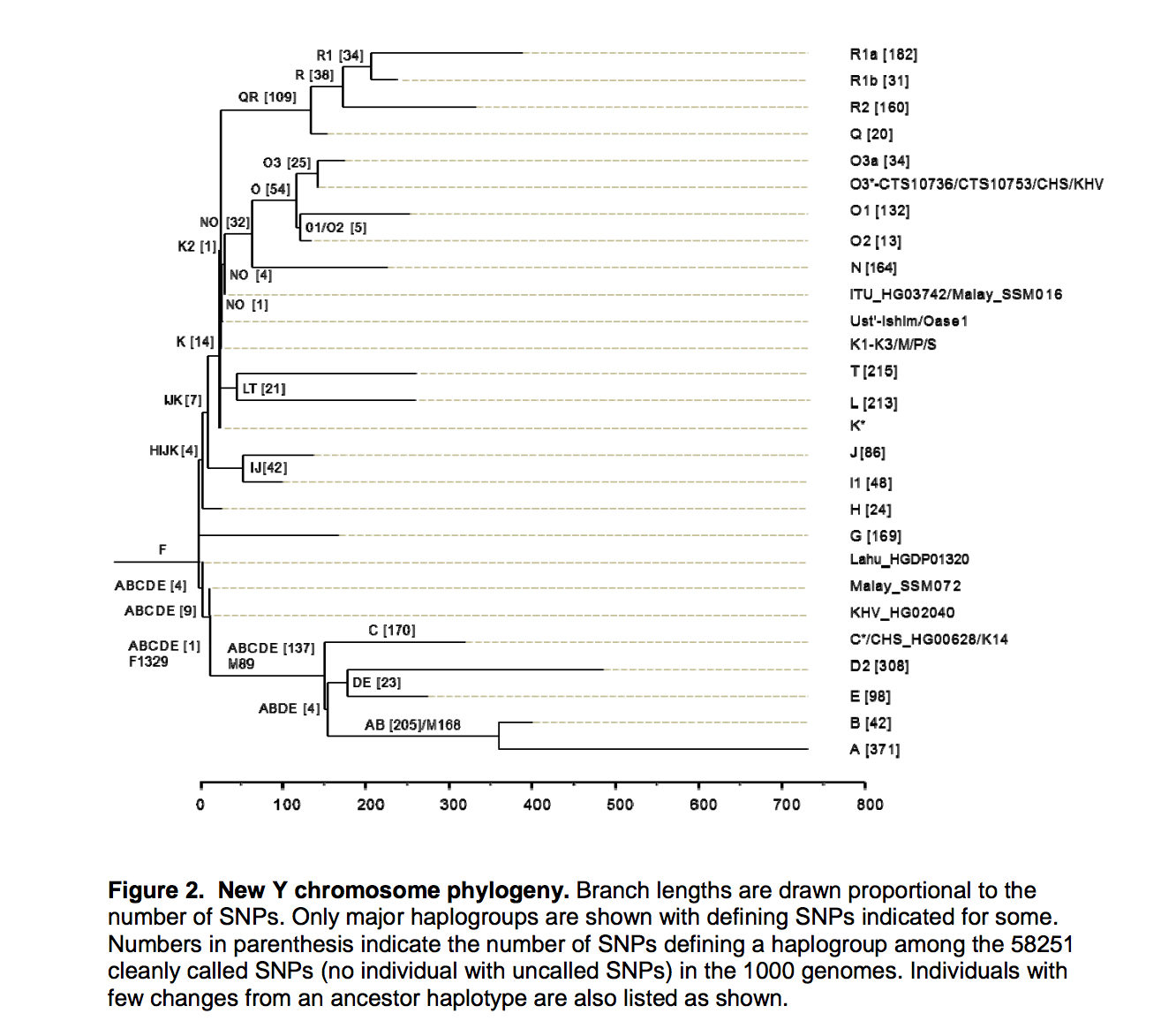
Y-DNA phylogeny from Yuan et al. (2017)
Y-DNA phylogeny from Poznik et al. (2013)
The difference between the two phylogenies involves two major changes. First, the divergent African hgs A and B are now subsumed under one single clade ABCDE of both-African and non-African distribution. They found increased allele sharing between ABCDE clade and Neanderthal Y chromosomes (see below).
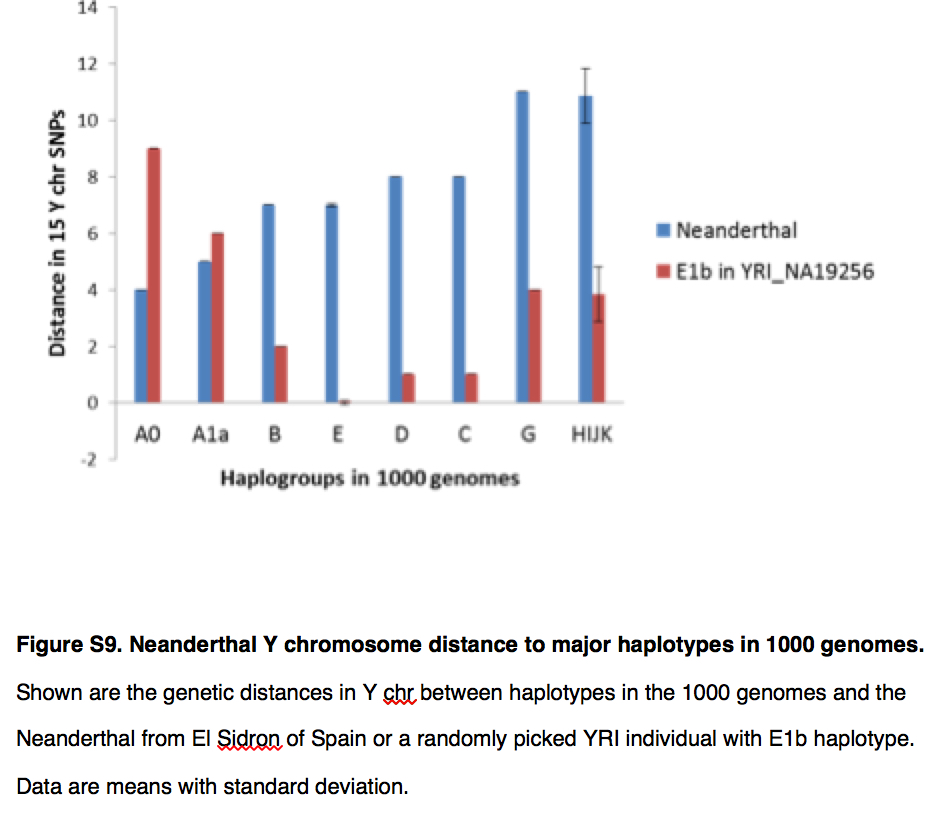
This sharing is especially pronounced for the African-specific hgs A0, A1a and B.
Second, ABCDE is a sister clade to hg F that encompasses the rest of the human Y-DNA lineages. Hg F is non-African. Overall, therefore, African haplogroups A, B and E are a subset of non-African clades. Y-DNA researchers have long argued that pan-African hg E marked by a very distinctive YAP+ mutation is nested within Eurasian variation and a back-migration from Asia to Africa was proposed (Altheide, T. K., and M. F. Hammer. “Evidence for a possible Asian origin of YAP+ Y chromosomes”. American Journal of Human Genetics 61, no. 2 (1997): 462-6; Chandrasekar, A. et al. “YAP insertion signature in South Asia.” Annals of Human Biology 34, no. 5 (2007): 582-6). A back-migration interpretation, however, was its own worst enemy because it effectively eliminated an important piece of evidence for the initial out-of-Africa migration and left the divergent African hgs A and B “locked” inside Africa. Yuan et al. (2017) straightened this situation out and eliminated the need for an old but ill-supported migration out of Africa.
Yuan et al. (2017) name haplogroup F as the most basal human haplogroup (Ust’-Ishim Y-DNA at 45,000 YBP is found to belong to hg NO, which is part of mhg F). And a direct implication for out-of-Asia follows:
“The F* haplotype is most common in East Asia, present in 5 of 7 (71.4%) Lahu males in Yunnan of South West China (Black et al., 2006), 10-15% of Han and other minority Chinese, and low percentages (<10%) in South Asians and French.”
mtDNA
According to Yuan et al. (2017), the current mtDNA phylogeny is plagued by the same cladistic errors as the Y-DNA phylogeny. They similarly revise it to reflect an out-of-Asia solution. First, the divergent African lineages L0, L1, L2, L4, L5 and L6 are subsumed under mhg L3. Second, hg L3 is shown to form a clade with a predominantly non-African mhg M. (Lippold et al. “Human paternal and maternal demographic histories: insights from high-resolution Y chromosome and mtDNA sequences.” Investigative Genetics 5 (2014), Fig. 3, also arrived at the grouping of hgs M and L3 into a single node opposed to hgs N and R.) Third, mhg R is posited as the basal modern human haplogroup from which both mhg M/L3 and mhg N are derived (see below).
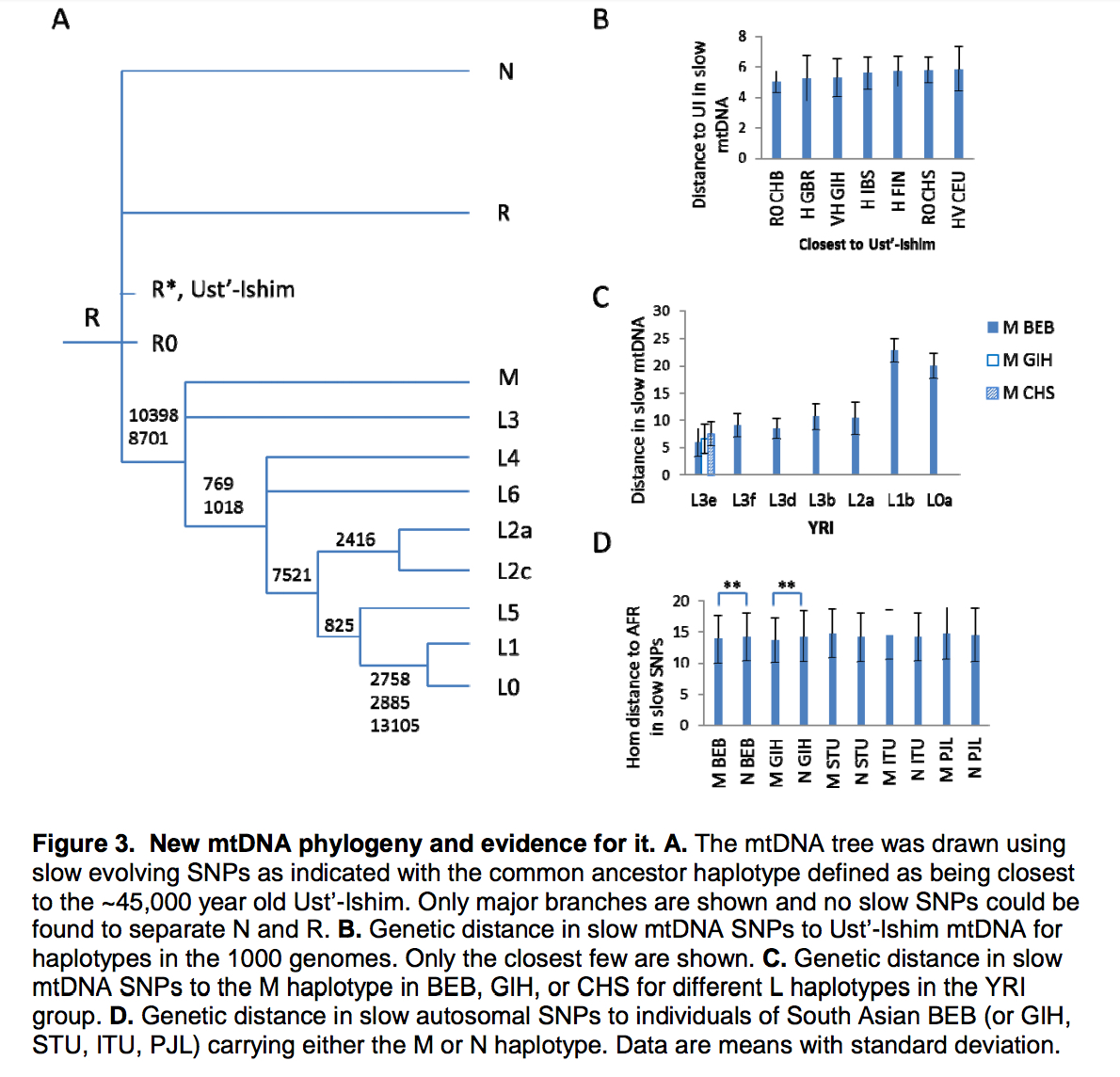
mtDNA phylogeny from Yuan et al. (2017)
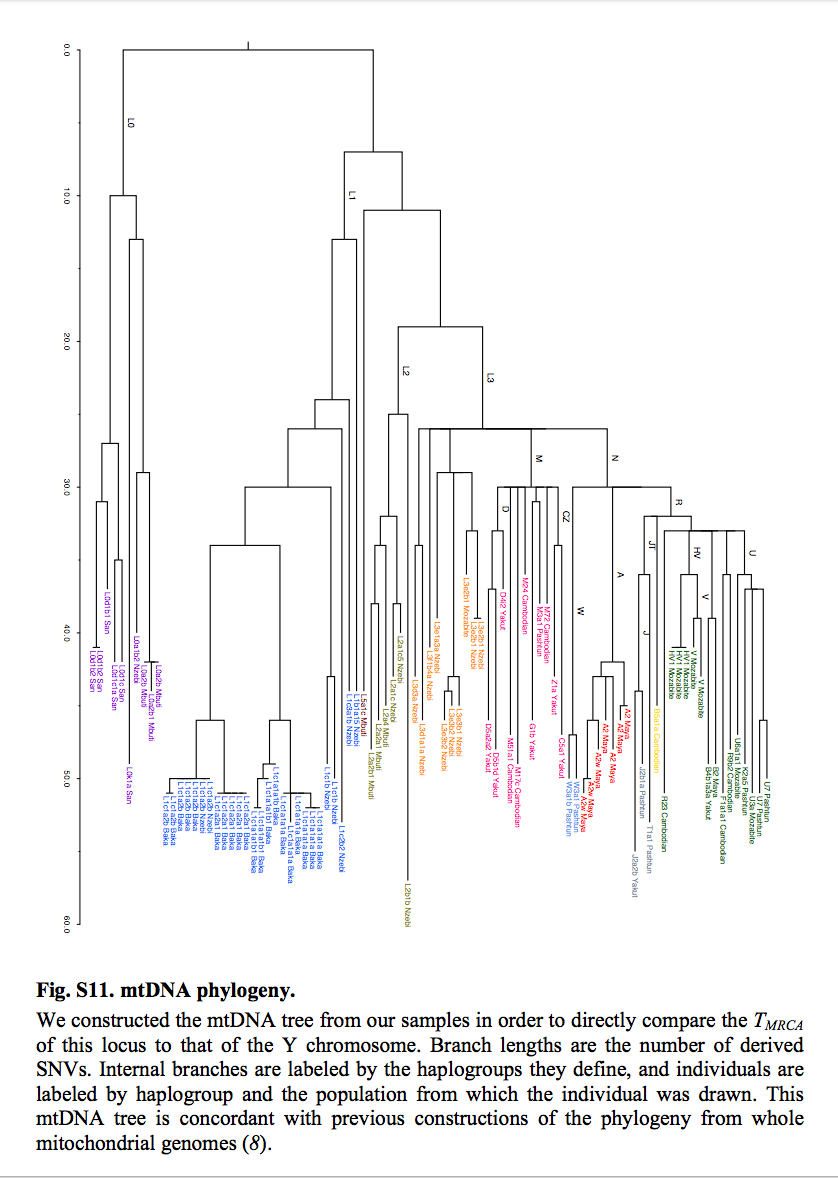
mtDNA phylogeny from Poznik et al. (2013)
This makes perfect sense considering that mhg R is the most widely spread human clade (its members are pervasive in America, Asia, the Sahul and West Eurasia). It’s also attested in all the most ancient DNA samples obtained to date: Ust’-Ishim (45,000 YBP), Tianyuan (40,000 YBP), Kostenki (32,000 YBP) and Mal’ta (24,000 YBP). Yuan et al.’s phylogeny revives the out-of-Asia phylogeny generated in a pioneering study by Johnson et al. (1983) from an early set of mtDNA morphs (see my analysis here). Johnson et al. used a “central types” method to root their tree. Unlike the “midpoint method” that assumed a constant mutation rate, the “central types” method favor mtDNA types that are geographically most widely spread and occur in more than one population. (Later, Cann et al. “Mitochondrial DNA and Human Evolution.” Nature 325 (187): 31-6) quietly walked away from the “central types” rooting method, excluded Amerindians from their sample and endorsed an out-of-Africa model under the assumption of a lineage-neutral molecular clock.)
An implication for out-of-Asia is presented in the following way:
“Two lines of evidence suggest haplogroup R as the ancestor of all modern haplogroups. First, ancient humans are expected to be closer to the ancestor and the oldest AMH, Ust’-Ishim, carried the R* haplotype (Fu et al., 2014). Second, R0 is the least differentiated haplotype and closest to the ancient haplotype in Ust’-Ishim (Figure 3B). That R0 is most common in Chinese among 1kGP indicates origin of R in East Asia (Figure 3B) and subsequent diversification in other regions of the world (Supplementary Figure S4). “
According to Gisele Horvat, the authors’ “hg R0” is in fact “B4”. One of these “R0” sequences is one of the B4k examples at Phylotree (NA18536, EU597559) (see below).

A specific example of consistency between mtDNA and Y-DNA trees, as redrawn by Yuan et al. (2017), involves Andamanese: they have Y-DNA hg D and mtDNA hg M. Yuan et al. (2017) argue for shared common ancestry between Africans (especially, Mbuti Pygmies), Andamanese and Philippine Negritos.
“The new Y tree grouping C with ABDE further suggests a common ancestry for different Negrito groups since the C haplotype is common in certain Negrito groups in Philippines while D is common in some others such as Onge.”
One could add that a large fraction of Austronesian (especially, Polynesian) Y chromosomes belong to hg C, which cannot be attributed to admixture with surrounding Papuan populations but have to go back to the original roots of Austronesians in Southeast Asia.
In a reversal of one of out-of-Africa models, namely the Southern Coastal model, they argue that Africa was colonized from Eurasia down a coastal route. This migration explains proximity between mtDNA hgs M and L3, on the one hand, and the shared Y-DNA clade membership of African hgs A, B and E with non-African hgs D and C. A coastal migration out of Africa never made sense because African mtDNA lineages were never reported from the Andaman islands, coastal Southeast Asia or from the Sahul. All the lineages from those areas were Asian, not African. A coastal migration from Asia to Africa, on the other hand, is a matter of course considering that a subset of mtDNA mhg M known as hg M1 has long been reported from East Africa and a “reverse” migration scenario was already inferred by other geneticists (see, e.g., Gonzalez et al. “Mitochondrial lineage M1 traces an early human backflow to Africa.” BMC Genomics 2007; 8: 223). By joining hgs M and L3’4’5’6’1’2 Yuan et al. simply deepened the impact of this known Asia-to-Africa migration on the African mtDNA landscape.
The reanalysis of mtDNA and Y-DNA data by Yuan et al. resolves the following long-standing problem with mtDNA and Y-DNA phylogenies: the most-widely shared alleles are most frequent in non-Africans but are deemed originating in Africa, while African-specific alleles are not found outside of Africa. Yuan et al. effectively put a nail in the coffin of the out-of-Africa theory. No more naive Afrocentrist musings about ancient African foragers, Khoisans and Pygmies, whose mothers diverged from the human Eve 140,000-70,000 years ago, who stayed in isolation until Holocene nomadic and agricultural expansions and who preserved ancient polyphonic music (Victor Grauer), first myths about the origin of death (Yuri Berezkin) as well as primitive phonetic sounds (clicks) (Knight et al. 2003). No more fantasies about the extreme time depths of lineage separation in Africa involving an enigmatic lineage A00, with very recent internal coalescence and easily found in African Americans, but somehow predating the divergence of all of non-African haplotypes by some 250,000 years.
At the same time, the new Y-DNA and mtDNA phylogenies explain some of the puzzling features of “cultural distributions”: polyphonic musical traits shared between African foragers, on the one hand, and a patchwork of Southeast Asian and Oceanian populations can be mapped back to the Y-DNA mhg ABCDE.
The Out-of-Asia Model of Modern Human Dispersals
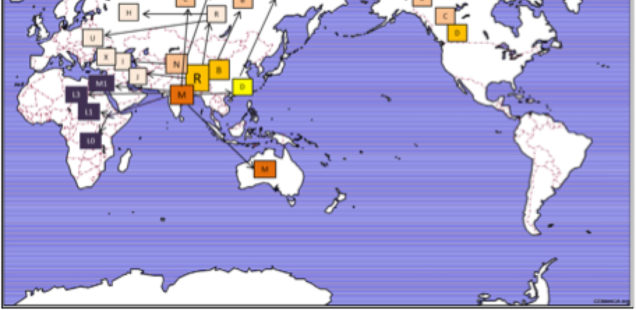
Yuan et al. (2017) summarize the implication of their genetic reanalysis to modern human origins and dispersals with the following map:
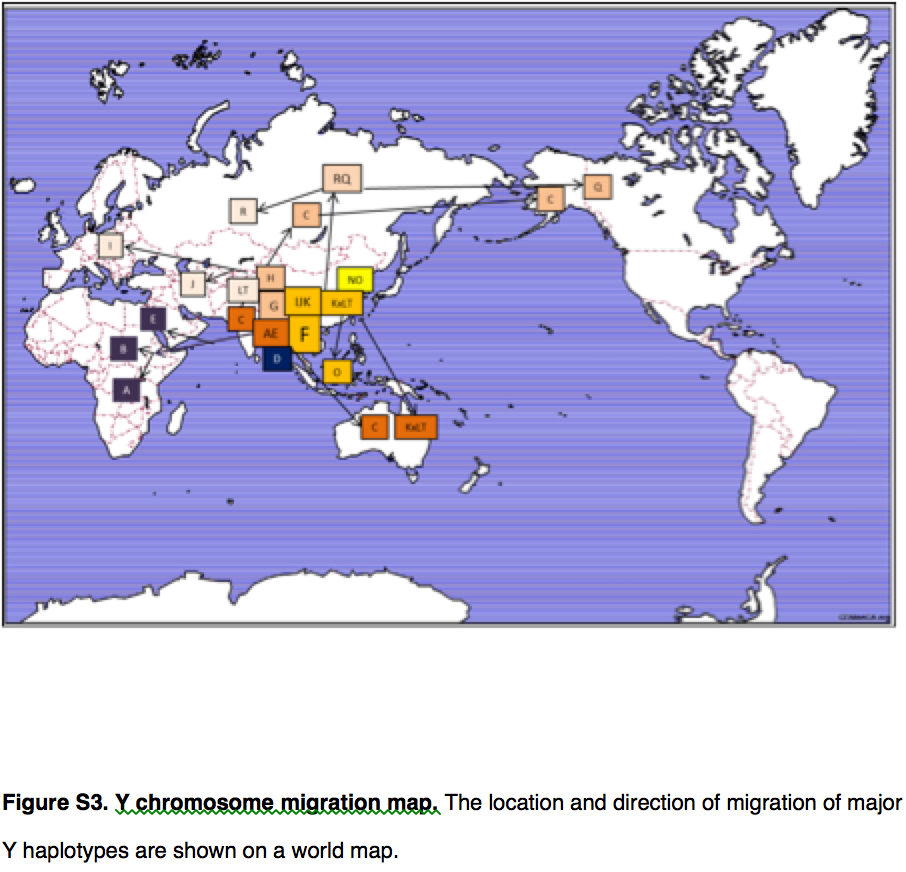
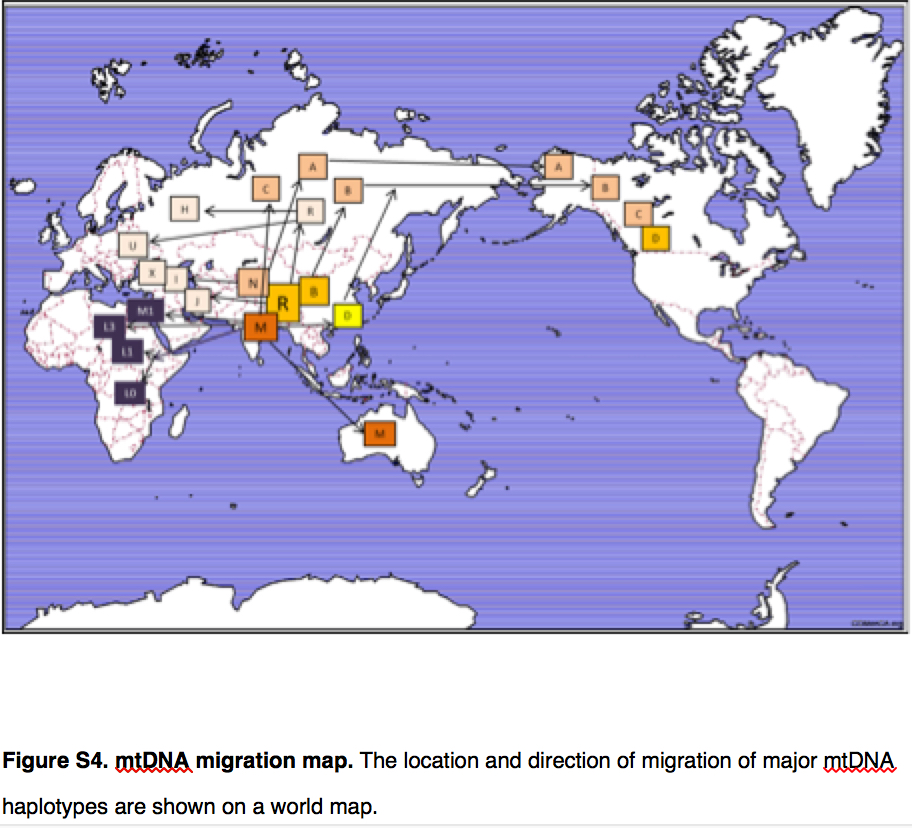
East Asia is the epicenter of modern human dispersals with two separate waves going westward toward Europe and Africa and eastward toward the Sahul and, across the Bering Strait, to the New World.
Out-of-Asia or Out-of-America?
Yuan et al. (2017) discuss in depth Africans, East Asians, Europeans, Australian aborigines, Negritos, Pygmies. They did not show that they had anything to say about the origins of Amerindians. If Amerindians are a relatively recent offshoot of East Asians, it remains unclear a) how did they acquire lineages from both ABCDE and F Y-DNA clades and from mtDNA mhg M’L3’4’5’6’1’2, mhg R and mhg N. Amerindian mtDNA hg B2 is phylogenetically very close to hg B4 (=R0) that Yuan et al. (2017) project as the oldest modern human lineage. Under the out-of-Asia theory and with much earlier dates for the peopling of the Americas (as suggested by Bluefish Caves), this phenomenon is easier to comprehend than under the out-of-Africa theory because America is geographically close to an epicenter of modern human dispersals in Asia. But it’s still an unexpected pattern; b) autosomal evidence is unambiguous regarding a special connection between Amerindians and West Eurasians (not shared by East Asians). This shared ancestry has an exact parallel in Y-DNA: the majority of West Eurasian and Amerindian Y chromosomes belong to sister hgs R and Q (part of mhg F). Significantly, ancient Ust’-Ishim and Oase Y-DNAs are hg NO, which is the most common haplogroup in modern Asians. But those haplogroups are entirely missing from America.
Some of findings suggest that out-of-America may be looming large behind Yuan et al.’s out-of-Asia. First of all, by showing the increasing presence of Neanderthal and Denisovan alleles with increasing proximity to Africa, Yuan et al. have removed some of the objections that have been raised against the out-of-America theory. Critics pondered why, if people moved across Eurasia into Africa, we don’t see hominin alleles in Africa. Yuan et al. have now showed that we do. Second, they write,
“We next tested certain obvious predictions of the out of East Asia model. First, the model predicts lower diversity in people directly associated with the original AMH and higher diversity in people resulting from admixture of AMH with archaic humans. We calculated the hom PGD in slow SNPs as well as het numbers for each of the 25 groups totaling 2534 individuals in 1kGP. The lowest hom PGD level was found in LWK followed by slightly higher level in CHS (Supplementary Figure S8A). However, LWK has significantly higher numbers of het than CHS (Supplementary Figure S8B). As high level heterozygosity indicates high genetic diversity and would reduce hom distance, it is likely that CHS has lower genetic diversity than LWK. We further found that within CHS (made of 72 individuals from Hunan and 36 from Fujian), Hunan samples have lower hom PGD and het numbers than Fujian samples (Supplementary Figure S8CD). These results indicate that CHS, in particular Hunan people, have lowest genetic diversity levels among the 25 groups in 1kGP. Given that known admixed groups such as MXL and PUR showed the highest genetic diversity or PGD (Supplementary Figure S8A), it may be inferred that CHS or Hunan people may have the least amount of admixture and hence represent the original AMH group, at least among the 25 groups sampled here.”
It’s rewarding to see geneticists actually using high homozygosity levels as an indicator of origins, not high heterozygosity levels – something that I’ve been arguing for for many years. But Yuan et al.’s own data cited above shows that Amerindians are even less diverse than East Asians, hence can be modeled as an even “purer” example of Homo sapiens sapiens. This is consistent with the lack of hominin fossils in the New World, on the one hand, and with the world-highest levels of linguistic diversity among Amerindians, on the other. Language and associated symbolic thinking remain key distinguishing traits of Homo sapiens sapiens, the ones that can easily explain the adaptive mechanisms behind their superior niche mastery and the replacement of archaic hominins.
Third, Yuan et al. use some famous “Asian” traits in African Khoisan (e.g., shovel-shaped incisors) as evidence of a migration from Asia to Sub-Saharan Africa.
“The much lower frequency of shoveling teeth in African fossils and Khoisan relative to ancient and modern Chinese suggests that this type of teeth could only originate in China with its African presence due to migration.”
But the very same trait is equally, if not more strongly expressed in Amerindians pointing therefore to an earlier, pre-East Asian onset of this frequency gradient prior to a population of expansion into Africa.
Fourth, the basal marker (type 1) in the out-of-Asia mtDNA phylogeny produced by Johnson et al. (1983), with which Yuan et al. (2017) phylogeny shares unmistakable similarities in assumptions about the mutation rate and in topology, was most frequent in Amerindians (see more here). This suggests that out-of-America may loom large behind out-of-Asia interpretations of modern human mtDNA evolution.



It all started in the glacial refuge on the Olympic Peninsula in Washington state. This I know.
The conclusions of the paper are contrary to a wealth of very solid scholarship by scores of scientists based upon on Y-DNA, mtDNA, autosomal genetics, and archaeological data.
It is unlikely to survive peer review and be accepted for publication.
Chinese scientists supported a multi-regional hypothesis largely for political reasons for many years, and a few seem to be returning to that path despite the fact that this hypothesis is widely discredited.
Your statements are meaningless (I began to refer to you at some point as a man of “encyclopedic ignorance” and this still holds) without a corresponding analysis of why one model is better supported by cross-disciplinary evidence than the other. As a dilettante in anthropology you can’t tell right from wrong, so you should just take a back seat, observe and ask questions. Using your ridiculous “Chinese jab,” I can satirically argue that Western science has built out-of-Africa out of a feeling of remorse for slave trade and to give support for its PC political agendas. Please refer to the Comments Policy for guidance of what and how to post on this blog.
Thank you for the reference to the comments policy. This is your space and I want to respect that and to post comments only on terms consistent with your policy.
I assume your main concern in my case is the portion that says: “Third, the commenter needs to bring value – this could be a smart, knowledgeable critique of my opinions, a creative elaboration of them or an interesting piece of data/news.”
Other portions of your policy pertain to pseudonym use. In this regard, I think that you are aware of this fact, but to be clear, the name that I use in posting comments is my real one (minus my first name, “Andrew” and hyphenation that computers tend to not parse well), and I have a linked website (Dispatches at Turtle Island). Obviously, as you indicate that you know, I am not a professional academic in these fields.
You got it right. Yes, your advantage over other commenters who often don’t get cleared is a real name and a linked website. Please avoid dogmatic rehashing of whatever you think is proven, mainstream ideas. Think of this blog as a startup launched by just another Stanford grad – don’t post about what a phone or a car are supposed to look like because this is what GM cars or Apple phones are. This is where new cars and new phones are being manufactured. So contribute info, ideas, links, critique, etc. Amateurs and pseudoanthropologists such as Maju, Razib, GCochran, Davidski or Dienekes are making a mistake of defining what the science of human origins is all about. They don’t know. This blog is one of the places where science is being done, not just dogmatically reported about.
Great news, the orthodoxy is being worn away by a more “open minded” theory, it is clear that Africa is not the cradle of mankind.
I generally like the research. A few points, in the name of Devil’s advocacy. I’ve read elsewhere that the Khoisan peoples of SW Africa are among the most ancient and primal peoples in all of Africa. They speak a click-language said to be extremely ancient. It’s a tough sell to say that their shovel-toothed incisors are an indication of a migration from East Asia. More likely, I would say, is that there was a migration from the Khoisan region to East Asia before the main migration out of Africa. And this early migration took hold strongly in China, but was wiped out in other parts of Asia by the later migration out of Africa.
Before the great expansion of the Bantu tribes, the Khoisan range was much larger, it covered all the way to Kenya and Tanzania.